La glomérulopathie à dépôts de C3 (GC3) et la glomérulonéphrite membrano-proliférative à complexes immuns (GNMP-CI) sont des maladies rénales rares et chroniques pouvant causer des dommages graves et irréversibles aux reins.1
Ces deux maladies sont des formes de glomérulonéphrite membrano-proliférative causées par un dépôt excessif et incontrôlé de fragments de la protéine C3 couplé à un dépôt d’immunoglobulines dans le cas de la GNMP-CI.2
Nombre de cas
L’incidence de la GC3 varie entre 0,2 et 2 cas par million par ans, avec une prévalence comprise entre 0,05 et 1,4 pour 10.000 individus.3,4
L’incidence combinée de la GC3 et de la GNMP-CI est comprise entre 1 à 5 cas par millions par ans.2,5
L’activation du complément dans la GC3 et la GNMP-CI
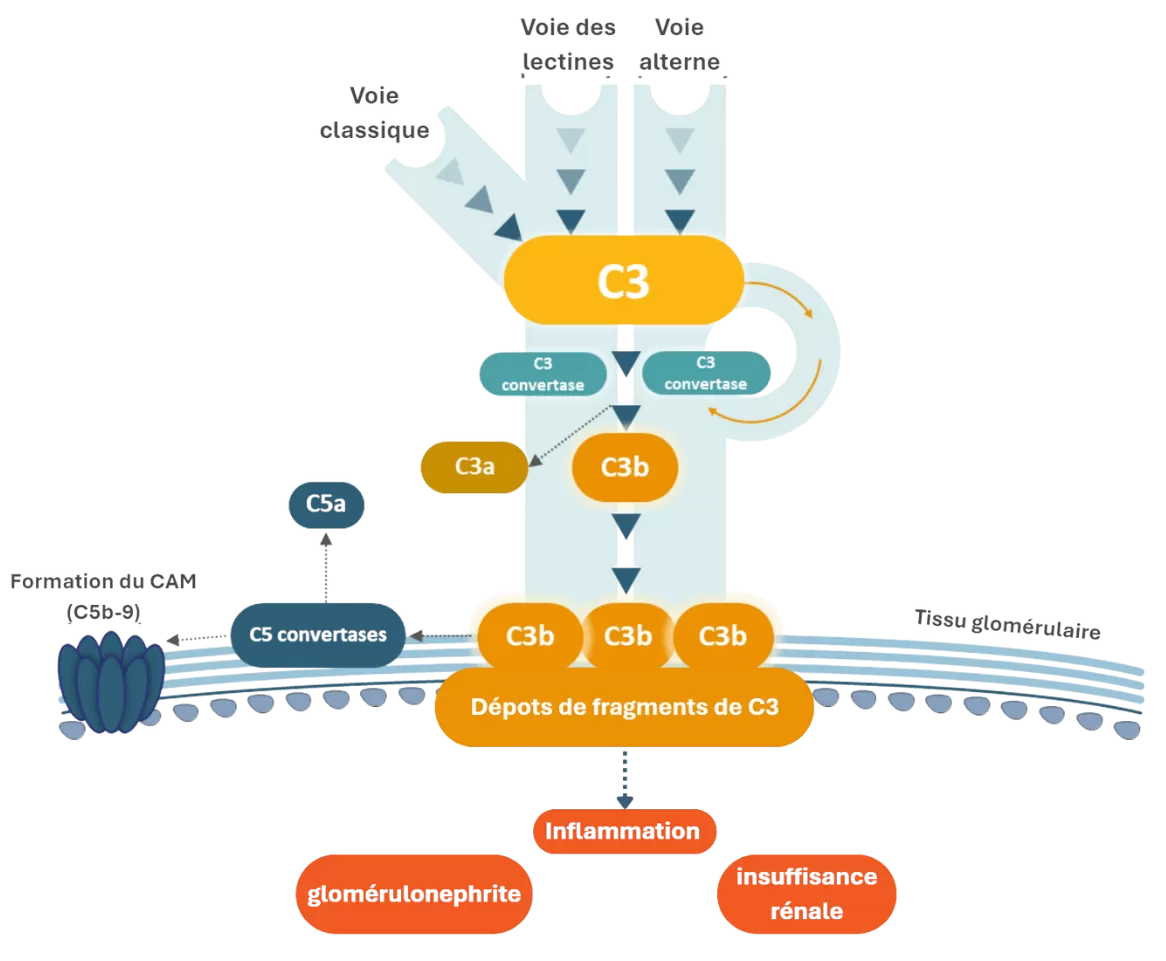
Bien que les mécanismes sous-jacents liés à la GC3 et à la GNMP-CI ne soient pas encore bien établis en raison de l’hétérogénéité de ces maladies, il existe un dénominateur commun entre ces dernières : la suractivation du complément.6
Le système du complément est un ensemble de protéines jouant un rôle clé dans l’immunité innée. Il participe à la destruction des agents infectieux et l’élimination des complexes immuns et des cellules apoptotiques.7
Dans la GC3, la voie alterne du complément est activée de manière incontrôlée en raison d’une dérégulation de la protéine C3 convertase. Cette dérégulation induit un dépôt et une accumulation des produits de dégradation de la protéine C3 dans les glomérules.6,7
Dans la GNMP-CI, la voie classique du complément est elle aussi activée de manière incontrôlée, entrainant des dépôts de complexes immuns. Toutefois, les mécanismes conduisant à ces dépôts sont encore mal compris.6,7,8
Cette accumulation de fragments de C3 dans le glomérule est associée au recrutement de cellules inflammatoires, perturbant la fonction membranaire et provoquant une inflammation, ou glomérulonéphrite.1,9
La glomérulonéphrite peut provoquer1, 10 :
- Des lésions glomérulaires continues
- Des ruptures de la membrane basale glomérulaire
- Des syndromes néphrotiques (protéinurie massive et œdème)
Diagnostic
La GNMP-CI est diagnostiquée à l’âge moyen de 20 ans. La GC3 est, elle, diagnostiquée à un âge supérieur (30 ans). Une prédominance masculine est constatée, avec 65 % de patients hommes pour la GNMP-CI et 59 % pour la GC3.6
La GC3 et la GNMP-CI peuvent être différenciées sur la base des résultats d'immunofluorescence réalisées sur des coupes de biopsies rénales8 :
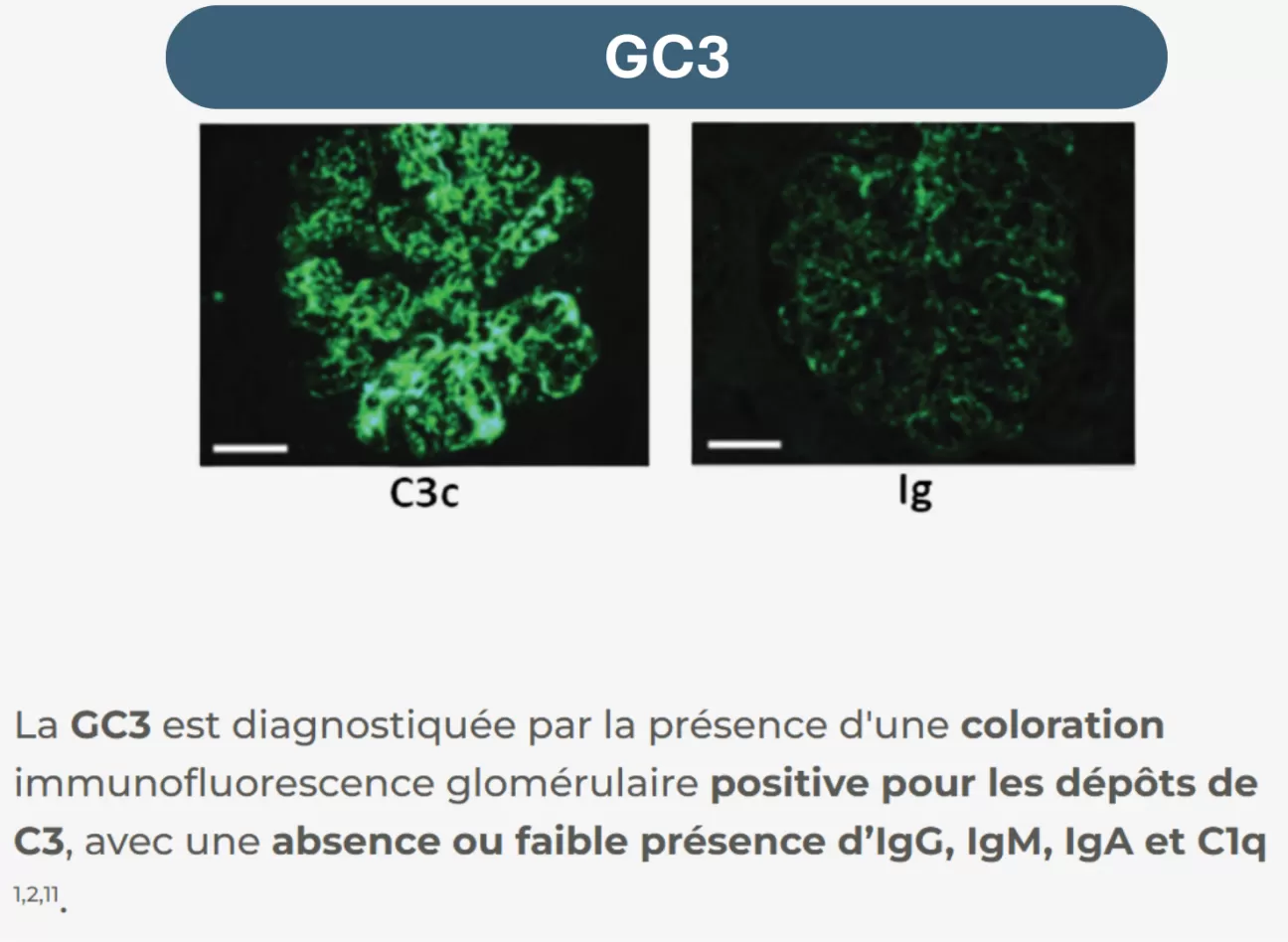
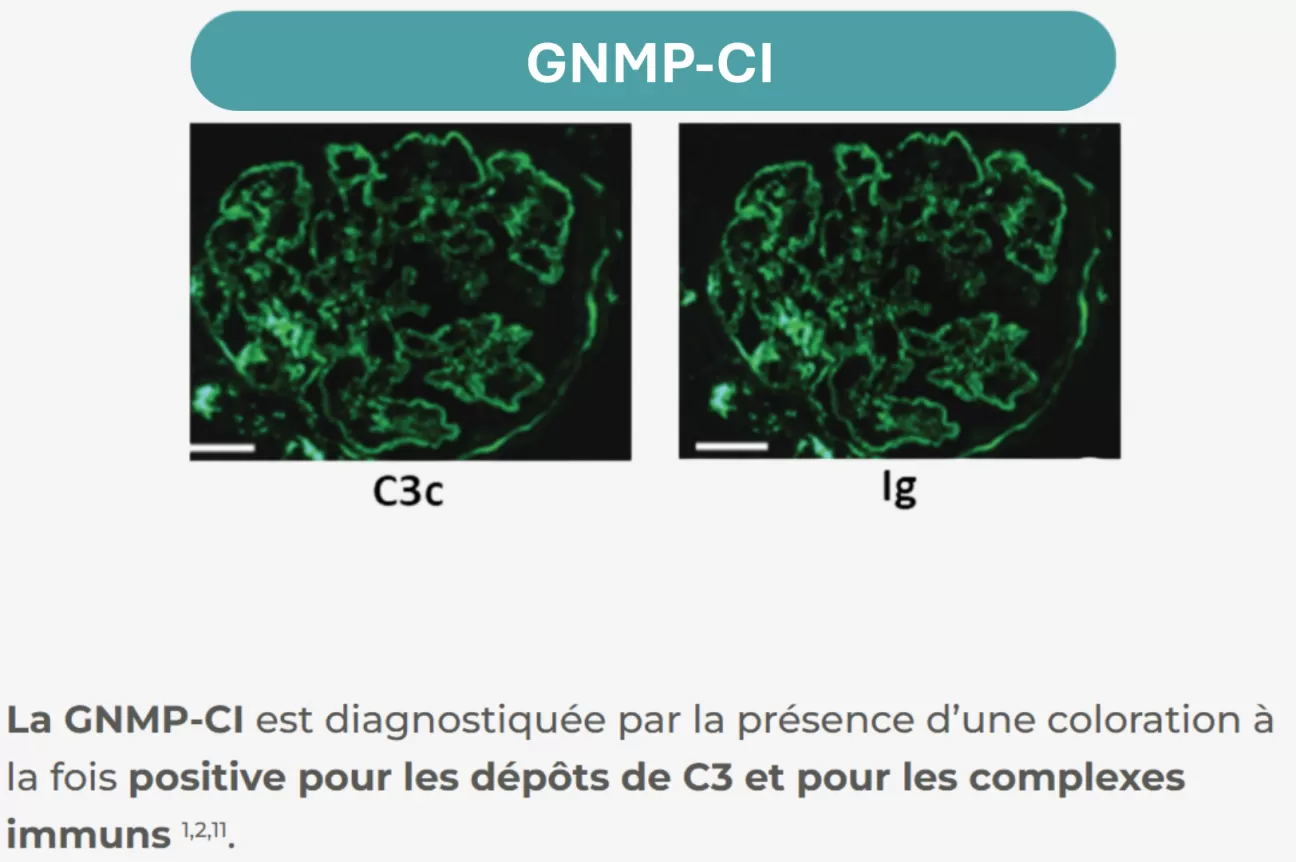
Dans la GNMP-CI, les dépôts d’immunoglobulines peuvent être provoqués par des conditions secondaires telles que des maladies infectieuses ou auto-immunes, des gammapathies monoclonales ou des dysprotéinémies. Lorsque toute condition secondaire est exclue, une GNMP-CI primitive est diagnostiquée12.
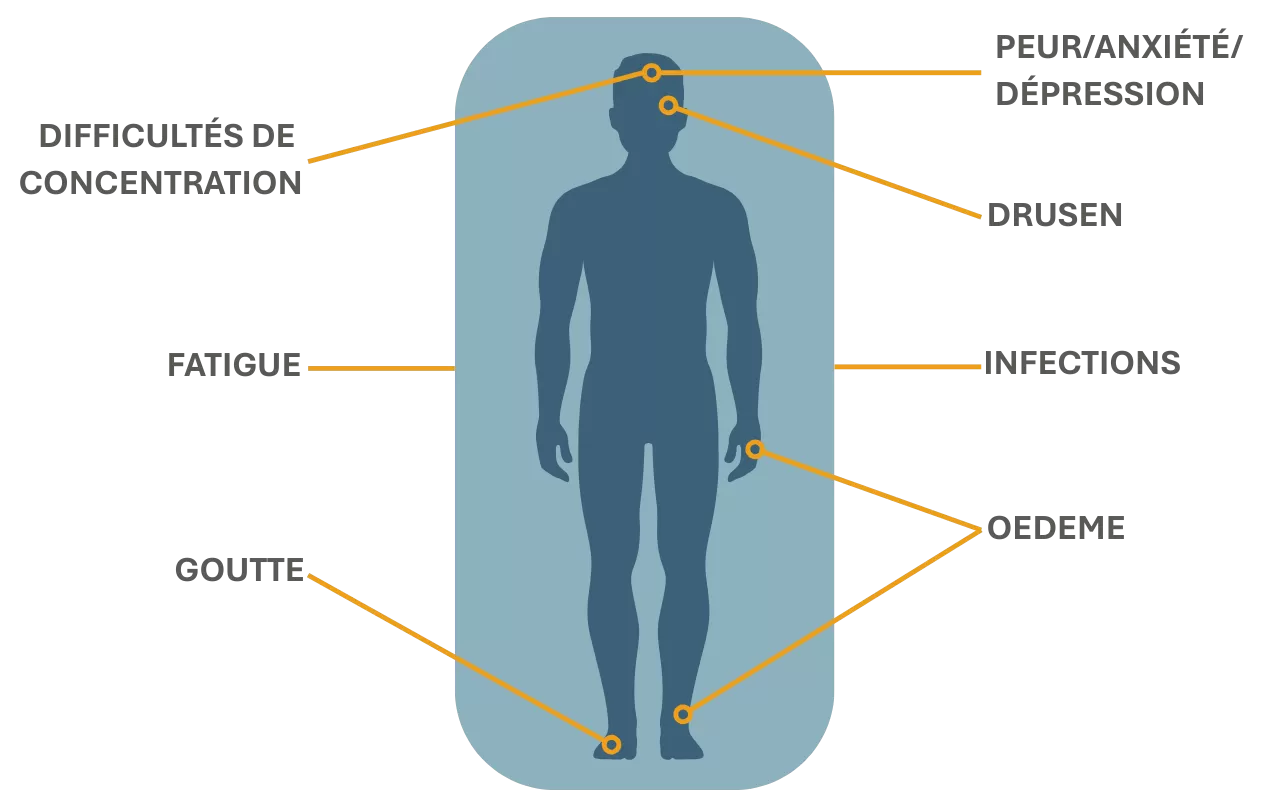
Manifestations cliniques et impacts sur les patients
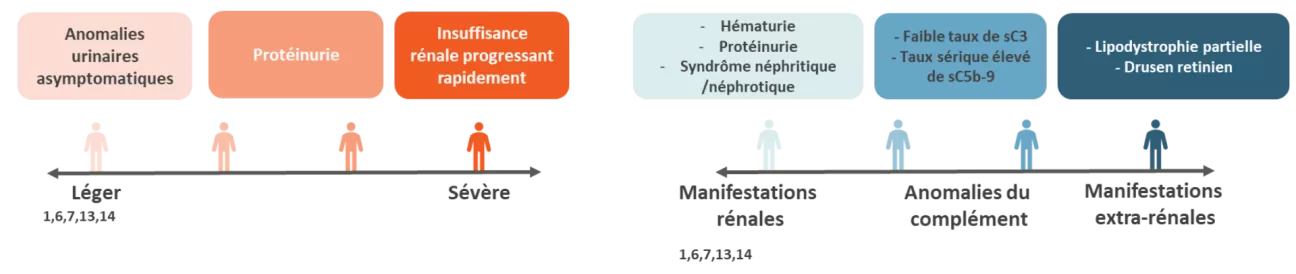
Les manifestations cliniques et biologiques de ces deux pathologies et leur état d’avancement au moment du diagnostic varient considérablement d’un patient à l’autre, rendant le diagnostic de ces maladies souvent long et imprécis.1,13
Ces manifestations cliniques se traduisent par des symptômes impactant grandement la qualité de vie des patients et limitant leurs activités au quotidien15
Pronostic et évolution
Malgré de grandes variations interindividuelles, l’évolution de la pathologie est souvent similaire : beaucoup de patients évolueront vers une insuffisance rénale terminale et nécessiteront des dialyses ou transplantations, ces dernières s’avérant souvent insuffisantes face à la récidive potentielle de la maladie.8,16,17,18
35-50 %
Des patients évoluent vers l'insuffisance rénale 5 à 10 ans après leur diagnostic1, 6, 7, 19
60-90 %
Des patients subissent une récidive de la maladie après une transplantation16, 17
43-60 %
Des patients subissent une perte de l'allogrèffe suite à une récidive de la maladie16, 18
De nombreux besoins restent en effet non couverts dans la prise en charge des patients atteints de GC3 et de GNMP-CI, notamment15 :
- L’accès à un diagnostic rapide et précis
- La prévention de la progression de la maladie vers l’insuffisance rénale
- La réduction du risque de transplantation et de la récurrence post-transplantation
- L’amélioration de la qualité de vie grâce à de nouvelles approches thérapeutiques innovantes
Références
1 Caravaca-FontánF, LucientesL, CaveroT, et al. Update on C3 Glomerulopathy: A Complement-MediatedDisease. Nephron. 2020;144(6):272-280.
2 Noris M, Remuzzi G. C3G and Ig-MPGN-treatment standard. Nephrol Dial Transplant. 2024;39(2):202-214.
3 Caravaca-FontánF, Toledo-Rojas R, Huerta A, et al. Comparative Analysis of Proteinuriaand Longitudinal Outcomesin Immune Complex MembranoproliferativeGlomerulonephritisand C3 Glomerulopathy. Kidney Int Rep. 2025;10(4):1223-1236.
4 Nester C, Decker DA, Meier M, et al. Developing Therapies for C3 Glomerulopathy: Report of the Kidney Health Initiative C3 Glomerulopathy Trial Endpoints Work Group. Clin J Am Soc Nephrol. 2024;19(9):1201-1208.
5 Masoud S, Wong K, Pitcher D, et al. Quantifying association of early proteinuria and estimated glomerular filtration rate changes with long-term kidney failure in C3 glomerulopathy and immune-complex membranoproliferative glomerulonephritis using the United Kingdom RaDaR Registry. Kidney Int. 2025;108(3):455-469.
6 Servais A, Noël LH, Roumenina LT, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82(4):454-64.
7 Smith RJH, Appel GB, Blom AM, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol. 2019;15(3):129-143.
8 Fakhouri F, Le Quintrec M, Frémeaux-Bacchi V. Practicalmanagement of C3 glomerulopathyand Ig-mediatedMPGN: factsand uncertainties. Kidney Int. 2020;98(5):1135-1148.
9 Schena FP, Esposito P, Rossini M. A Narrative Review on C3 Glomerulopathy: A Rare Renal Disease. Int J Mol Sci. 2020;21(2):525.
10 Anders HJ, KitchingAR, Leung N, et al. Glomerulonephritis: immunopathogenesisand immunotherapy. Nat RevImmunol. 2023;23(7):453-471.
11 Michels MAHM, Volokhina EB, van de Kar NCAJ, et al. Challenges in diagnostic testing of nephritic factors. Front Immunol. 2022;13:1036136.
12 Fervenza FC, Sethi S, Glassock RJ. Idiopathic membranoproliferative glomerulonephritis: does it exist? Nephrol Dial Transplant. 2012;27(12):4288-94.
13 Politano SA, Colbert GB, Hamiduzzaman N. Nephrotic Syndrome. Prim Care. 2020;47(4):597-613.
14 BombackAS, SantorielloD, AvasareRS, et al. C3 glomerulonephritisand dense depositdisease sharea similardisease course in a large United States cohortof patients with C3 glomerulopathy. Kidney Int. 2018;93(4):977-985.
15 Feldman DL, Bomback A, Nester C. Voice of the patient. Report of Externally-led Patient-Focused Drug Development Meeting on: Complement 3 Glomerulopathy (C3G). 2018
16 Caravaca-FontánF, PolancoN, VillacortaB, et al. Recurrenceof immune complex and complement-mediatedmembranoproliferativeglomerulonephritisin kidney transplantation. NephrolDial Transplant. 2023;38(1):222-235.
17 Noris M, Daina E, Remuzzi G. Membranoproliferative glomerulonephritis: no longer the same disease and may need very different treatment. Nephrol Dial Transplant. 2023;38(2):283-290.
18 Tarragón B, Peleg Y, Jagannathan G, et al. C3 Glomerulopathy Recurs Early after Kidney Transplantation in Serial Biopsies Performed within the First 2 Years after Transplantation. Clin J Am Soc Nephrol. 2024;19(8):1005-1015.
19 Lomax-Browne HJ, Medjeral-Thomas NR, Barbour SJ, et al. Association of Histologic Parameters with Outcome in C3 Glomerulopathy and Idiopathic Immunoglobulin-Associated Membranoproliferative Glomerulonephritis. Clin J Am Soc Nephrol. 2022;17(7):994-1007.
Janv., 2026