La tyrosinémie héréditaire de type 1 (HT-1) est une maladie génétique rare et grave au cours de laquelle l'organisme n’arrive pas à dégrader complètement un acide aminé appelé tyrosine. Elle est due à une mutation au niveau du gène codant la FAH (Fumaryl Acétoacétate Hydrolase ou Fumarylacétoacétase) qui est impliqué dans la voie de dégradation de la tyrosine. (1)
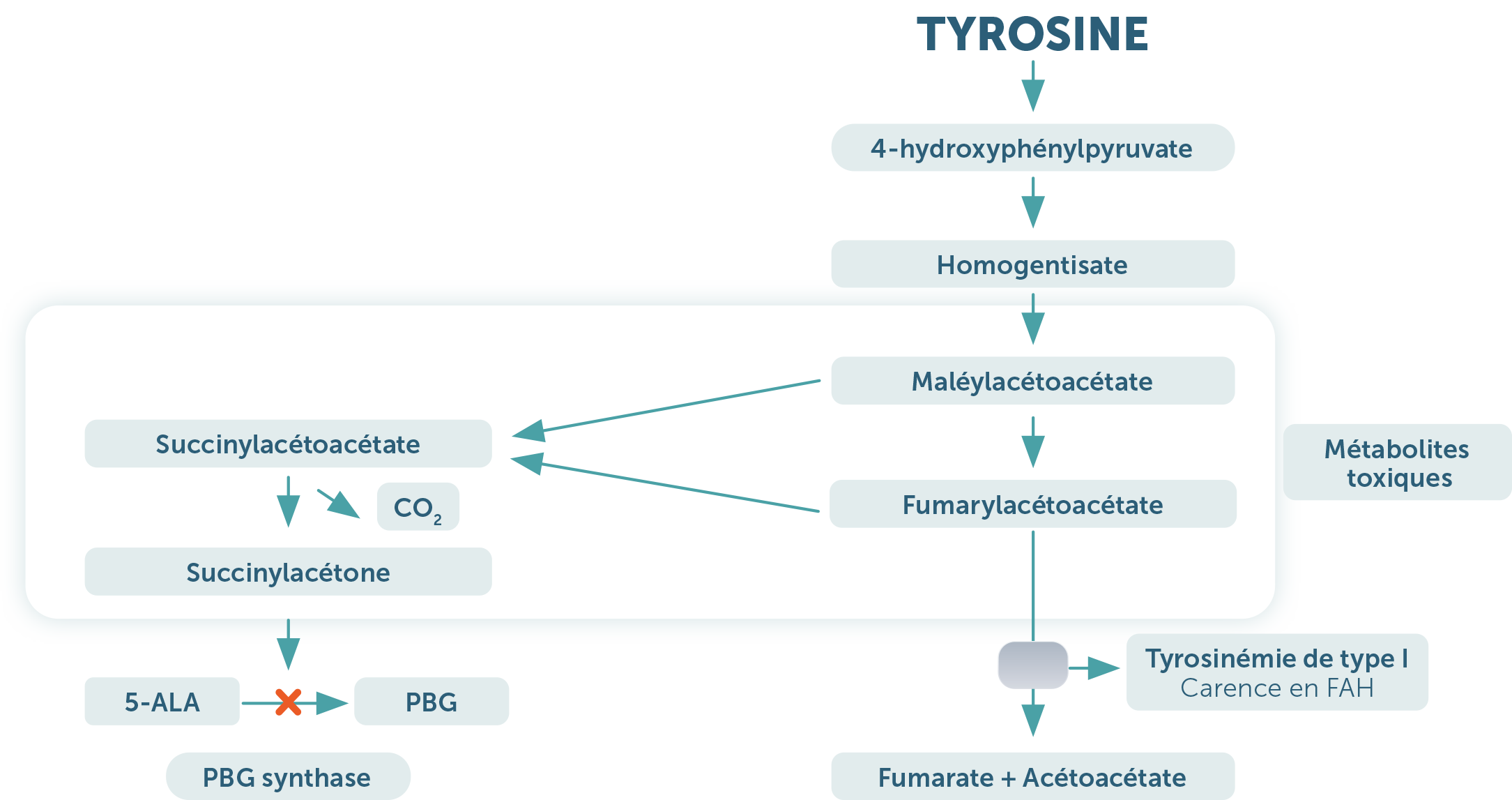
Voie catabolique de la tyrosine et HT-1 (1)
La conséquence de ce déficit est l'accumulation de substances toxiques dans le sang pouvant causer des atteintes au niveau du foie, un dysfonctionnement rénal et des troubles neurologiques. Sans prise en charge thérapeutique et diététique, le pronostic vital est engagé.
À l'échelle mondiale, la tyrosinémie de type 1 touche environ un nouveau-né sur 100 000 ; dans certains pays, notamment au Québec dans la région du Saguenay-Lac-St-Jean, on retrouve cette maladie avec une prévalence plus élevée (2). Une généalogie consanguine pourrait expliquer ces pics de prévalence.
Il existe 3 formes cliniques d’HT1, dépendantes de l’âge de survenue des premiers symptômes, et qui sont corrélées avec la sévérité de la maladie (1)(3) :
- Forme aiguë (apparition de la maladie avant l’âge de 6 mois).
C’est la forme la plus fréquente. Les patients développent en général des signes de problèmes du foie dans les premières semaines ou les premiers mois de vie, associant un retard de croissance, des vomissements, une diarrhée, une fièvre, parfois des selles noires et des saignements de nez. La forme la plus grave apparaît entre 0 et 2 mois.
- Forme sub-aiguë (apparition de la maladie entre 6 mois et 1 an).
La survenue des symptômes est moins rapide et l’expression moins sévère. Les symptômes principaux sont : des problèmes du foie, des reins, et de la coagulation, un retard de croissance, et une baisse du tonus musculaire.
- Forme chronique (apparition de la maladie chez les patients de plus de 1 an).
La progression de la maladie est lente. Cette forme se caractérise par une insuffisance hépatique graduelle, et provoque fréquemment des cancers du foie. En outre, cette forme d’HT1 est caractérisée notamment par la présence d’une cirrhose, des problèmes cardiaques, rénaux et neurologiques.

Principales manifestations cliniques en fonction de la forme associée (1)(3)
Diagnostic de la tyrosinémie héréditaire de type 1 (1)(3)
Concernant le diagnostic, il existe un test spécifique consistant à mesurer le taux d’une substance, appelée succinylacétone (SA), retrouvé dans le sang ou les urines.
Quand les futurs parents d’enfants sont atteints de cette maladie, il est possible de réaliser ce que l’on appelle un diagnostic prénatal, en mesurant le taux de SA dans le liquide amniotique. Un diagnostic néonatal peut également être réalisé chez des nouveaux-nés pour lesquels la maladie ne s’est pas encore manifestée.
Prise en charge de la tyrosinémie héréditaire de type 1 (1)
Historiquement, la prise en charge reposait sur un régime diététique limité en tyrosine et en phénylalanine, avec ou sans transplantation hépatique. La transplantation hépatique présente un taux de morbi-mortalité s’élevant approximativement entre 5-10%.
Aujourd’hui, grâce au diagnostic précoce de la maladie, une prise en charge thérapeutique spécifique peut être mise en place. Depuis le début des années 90, cette prise en charge consiste en un traitement médicamenteux quotidien associé obligatoirement à un régime alimentaire spécifique contrôlé en protéines naturelles, avec une consommation journalière d’acides aminés sans phénylalanine et tyrosine.
Références
(1) Fernandes J et al. Inborn Metabolic Diseases 4th Edition. Springer 2006 - chap18: pages 235 à 238
(2) De Braekeleer M et al. Genetic epidemiology of hereditary tyrosinemia in Quebec and in Saguenay-Lac-St-Jean. Am J Hum Genet. 1990 Aug;47(2):302-7.
(3) De Laet C, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013 Jan 11;8:8. doi: 10.1186/1750-1172-8-8. Review.
Août, 2025