La fièvre méditerranéenne familiale (FMF) est une maladie auto-inflammatoire rare. Elle peut se manifester par de la fièvre accompagnée de douleurs abdominales, articulaires ou thoraciques. La FMF est causée par des mutations du gène MEFV (pour MEditerranean FeVer), transmis d’une génération à la suivante.
Qu’est-ce que la FMF ?
La FMF est une maladie auto-inflammatoire héréditaire qui touche en particulier les populations originaires du pourtour méditerranéen ou du Moyen-Orient.1,2 Elle se caractérise par des accès répétés de fièvre et d’inflammation douloureuse au niveau de l’abdomen, du thorax et des articulations.
La FMF est causée par des mutations (modifications) du gène MEFV codant pour une protéine, la pyrine, qui aide à réguler le système immunitaire. Chez les personnes atteintes de FMF, des mutations du gène MEFV sont à l’origine d’une réponse excessive du système immunitaire à des stimuli qui ne causent habituellement pas d’inflammation. On observe alors des épisodes fiévreux et douloureux.3,4
Les symptômes de la FMF apparaissent généralement dans l’enfance ou à l’adolescence. Dans 80 % des cas, les premiers symptômes de FMF se développent avant l’âge de 20 ans.5 Un contrôle insuffisant de l’inflammation chronique peut conduire à des complications graves telles qu'une amylose rénale AA, susceptible d’aboutir à une insuffisance rénale.6 Bien que la FMF soit une maladie chronique nécessitant un traitement à vie, les patients peuvent contrôler efficacement leurs symptômes avec un traitement adéquat, et le pronostic à long terme est généralement bon.7,8,9
Maladies auto-inflammatoires et auto-immunes : quelle est la différence ?
Les maladies auto-inflammatoires et auto-immunes sont deux types différents de troubles du système immunitaire.
Les maladies auto-inflammatoires causent une inflammation pouvant survenir en l’absence de déclencheur ou d’infection évidents. Ces affections sont causées par des mutations génétiques touchant le système immunitaire inné.10 Les maladies auto-inflammatoires se caractérisent par une activation inappropriée du système immunitaire, à l’origine d’une inflammation excessive et de lésions tissulaires. La fièvre méditerranéenne familiale est le syndrome auto-inflammatoire le plus fréquent.11
Les maladies auto-immunes surviennent lorsque le système immunitaire attaque par erreur les propres tissus sains de l’organisme, qu’il considère comme étrangers.12 Ces maladies peuvent toucher toutes les régions de l’organisme et leurs symptômes sont très variés. La thrombopénie immune (TPI) ou purpura thrombopénique immunologique (PTI) et l’hémoglobinurie paroxystique nocturne (HPN), par exemple, sont des maladies auto-immunes.
Quelle est la fréquence de la FMF ?
Bien que rare dans la plupart des régions du monde, la FMF est la maladie auto-inflammatoire héréditaire la plus fréquente. La maladie touche le plus souvent les Juifs séfarades, les Arméniens, les Turques, les Arabes et les Marocains – dont l’origine familiale peut être identifiée dans le bassin méditerranéen.13 Tous les groupes ethniques peuvent être atteints. La maladie est considérée comme rare, même si sa fréquence est bien plus élevée au sein de ces populations méditerranéennes et peut ainsi atteindre 1 personne sur 200.14 La FMF n’est pas considérée comme rare en Turquie, en Israël et en Arménie.
Dans certaines populations juives, la proportion de personnes porteuses du gène muté, mais non atteintes, peut atteindre 6 % à 39 %.15
La fréquence de la FMF est à peu près équivalente entre hommes et femmes.16
Quels sont les symptômes de la FMF ?
Les symptômes peuvent être des douleurs abdominales ou thoraciques, un gonflement et des douleurs articulaires (arthralgies), de la fièvre, des éruptions cutanées et des céphalées.17
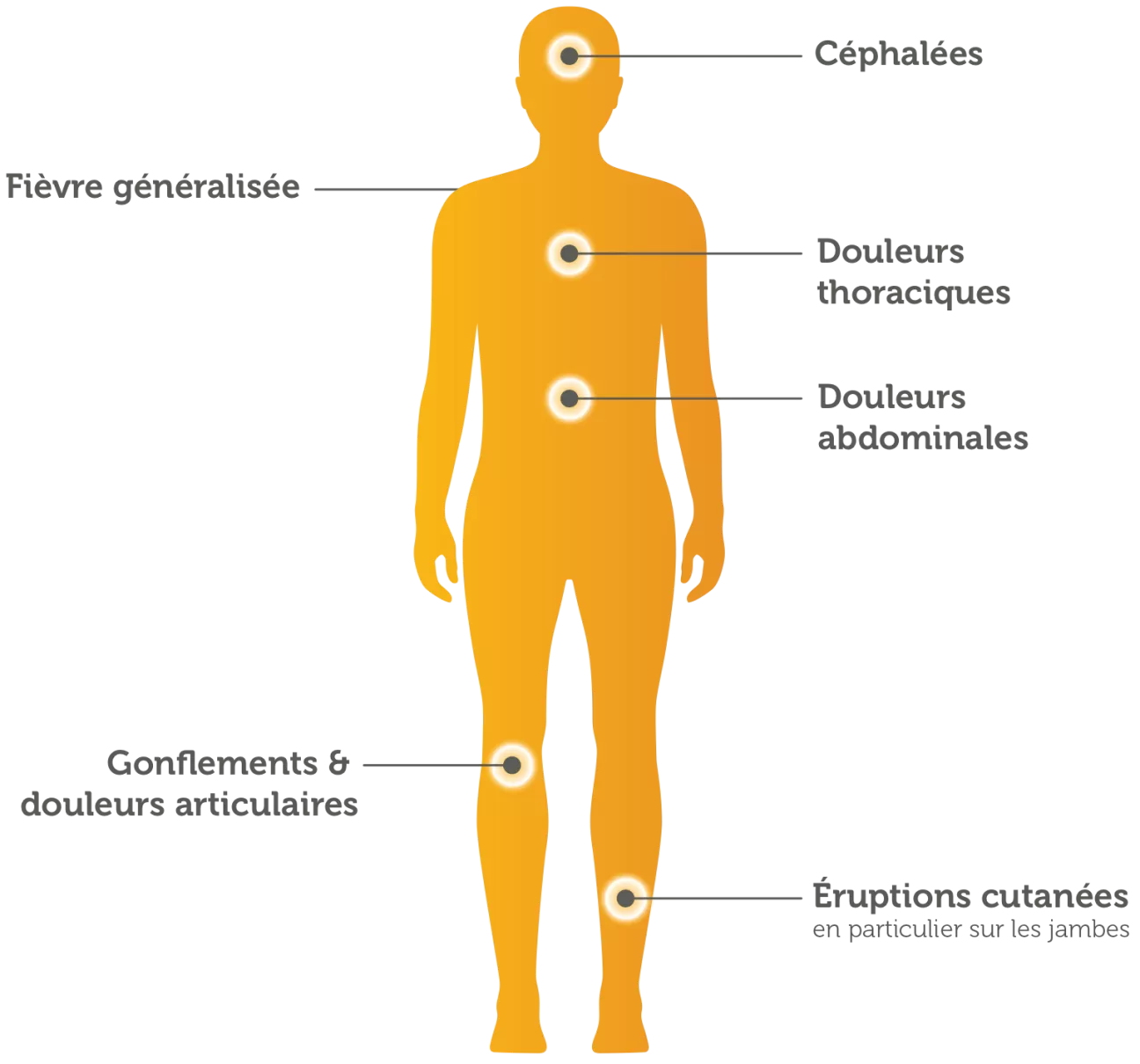
Dans la plupart des cas, la première crise est observée dans l’enfance ou à la puberté. Elle dure généralement deux à trois jours. La fréquence des crises est variable. Elles peuvent être espacées de seulement quelques jours à plusieurs années. Environ la moitié des personnes atteintes par la maladie ressentent une légère gêne (prodromes) avant le début de la crise.18
Complications possibles de la FMF
En l’absence de traitement, des dépôts de protéines, appelés amyloïdes, peuvent s’accumuler dans les organes et tissus de tout l’organisme. L’amylose désigne un groupe d’affections rares, graves causées par l’accumulation de dépôts amyloïdes. L’amylose touche en particulier les reins et peut aboutir à une insuffisance rénale.19
L’amylose peut aussi atteindre les testicules et les ovaires, causant potentiellement une infertilité chez l’homme et la femme.20
Comment diagnostiquer la FMF ?
Le diagnostic de FMF est complexe car les symptômes sont parfois similaires à ceux d’autres troubles inflammatoires. Le médecin cherche une fièvre et une inflammation répétées associées à d’autres symptômes – douleurs abdominales, douleurs thoraciques, arthralgies et éruptions cutanées. Il s’agit des critères diagnostiques de la fièvre méditerranéenne familiale.21
Complexité du diagnostic
La fièvre familiale méditerranéenne étant une maladie rare dans de nombreux pays, certains prestataires de santé la méconnaissent ou ne la considèrent pas comme possible. La réalisation d’analyses appropriées ou l’orientation vers des soins spécialisés peuvent ainsi être retardées. Le diagnostic peut également être erroné. Par exemple, le premier accès soudain de douleurs abdominales d'un patient est parfois confondu avec une appendicite.22
Tests génétiques
En cas de suspicion de FMF, il est possible de pratiquer un examen génétique pour le diagnostic. Les tests génétiques nécessitent un prélèvement sanguin et l’analyse de l’ADN pour identifier des mutations des gènes associés à la FMF. Un résultat positif à l’analyse génétique, associé à une symptomatologie récurrente, peut confirmer un diagnostic de FMF.23,24
Quelles sont les causes de la FMF ?
La fièvre méditerranéenne familiale est causée par des mutations (modifications) du gène MEFV, qui contient les instructions nécessaires à la fabrication d'une protéine appelée pyrine. La pyrine aide à réguler la réponse inflammatoire de l’organisme en contrôlant la production de certaines protéines qui favorisent l’inflammation.25
Chez les patients atteints de FMF, les mutations du gène MEFV induisent la formation d'une pyrine non fonctionnelle, et par conséquent une inflammation excessive dans l’organisme. Cette inflammation peut causer des lésions de différents tissus et organes. 26
La FMF n’est pas contagieuse. Elle n’est pas non plus considérée comme idiopathique – c’est-à-dire qu’elle ne survient pas spontanément. La FMF est héréditaire, et non acquise. Le mode de transmission de la maladie est essentiellement autosomique, récessif.
Qu’est-ce que la transmission autosomique récessive ?
- Récessive signifie qu’il est possible qu’une personne soit porteuse, sans présenter de symptômes.
- Autosomique signifie que le gène n’est pas situé sur un chromosome sexuel. Les hommes et femmes ont donc le même risque d’être atteints (ou d’être porteurs)..27
En règle générale, seule une personne dont les deux parents sont porteurs de la mutation responsable de la FMF peuvent développer la FMF. La mutation peut se transmettre sur plusieurs générations, des deux côtés de la famille, avant qu’un enfant naisse avec la maladie.
Les crises de FMF peuvent survenir sans facteur déclenchant identifiable. Dans certains cas, des facteurs ont toutefois été identifiés. Les facteurs susceptibles de déclencher une crise sont les suivants : infection, traumatisme, activité physique intense, stress et fatigue. Chez la femme, le début des menstruations peut déclencher une crise.28
Facteurs de risque
L’origine ethnique et les antécédents familiaux sont les principaux facteurs de risque. 29,30
Origine ethnique – la maladie sévit essentiellement au sein de groupes ethniques méditerranéens – elle touche les personnes dont la famille est originaire du bassin méditerranéen.
Antécédents familiaux – la FMF étant héréditaire, le risque de développer la FMF est plus important chez les personnes dont certains membres de la famille proche ont la maladie.
Qu’est-ce que la pyrine ?
Les scientifiques n’ont pas totalement élucidé le mécanisme d’action de la pyrine. Elle contribuerait à contrôler le processus inflammatoire. L’inflammation se développe lorsque le système immunitaire envoie des molécules particulières et des globules blancs vers une zone lésée ou malade pour lutter contre des envahisseurs nocifs et favoriser la cicatrisation. Une fois cette tâche achevée, un organisme sain interrompt la réponse inflammatoire et évite ainsi les lésions de ses propres cellules et tissus. Chez une personne atteinte de FMF, la pyrine ne fonctionne pas normalement et la réponse inflammatoire peut persister.31
Ralentir ou interrompre les réponses inflammatoires
La pyrine est synthétisée dans certains types de globules blancs, impliqués dans l’inflammation et la lutte contre les infections. La pyrine semble guider ces globules blancs vers les sites d’inflammation. Elle ralentit ou stoppe ensuite la réponse inflammatoire lorsqu’elle n’est plus nécessaire. La pyrine interagit également avec d’autres molécules pour former des structures appelées inflammasomes, qui participent au processus inflammatoire.
Selon certaines études, la pyrine aiderait à réguler l’inflammation en interagissant avec la structure de soutien de la cellule. Cette structure, appelée cytosquelette, contribue à la forme, la taille et la mobilité de la cellule.
La pyrine est également appelée marénostrine.32
Quelles sont les prises en charge disponibles contre la FMF ?
On ne peut pas guérir la FMF. Cette maladie peut toutefois être traitée efficacement, une fois diagnostiquée.33 Les traitements de la FMF sont des médicaments qui réduisent l’inflammation et aident à contrôler les symptômes. 34,35,36
Comment mieux vivre avec la FMF ?
Un diagnostic et un traitement précoces aident la plupart des patients atteints de FMF à mener une vie normale.37
Alimentation, activité physique, contrôle du poids, lutte contre le stress – et traitement
Adopter un mode de vie sain, grâce à une bonne alimentation, de l’activité physique, un contrôle du poids et la lutte contre le stress, peut aider les patients à gérer leurs douleurs et à préserver leur santé.38 Il existe des médicaments qui permettent non seulement de traiter mais aussi de prévenir les symptômes dans de nombreux cas. 39,40,41
FMF et alimentation42
Aucun aliment en particulier ne déclenche les accès de FMF. Toutefois, certaines personnes atteintes de FMF ont identifié des facteurs pouvant déclencher des crises ou sont sensibles à certains aliments. Les aliments pouvant potentiellement déclencher des crises de FMF chez certaines personnes sont notamment des aliments gras et salés, le lait de vache, le lait maternisé et le blé.
La tenue d’un journal alimentaire, en surveillant les effets de différents aliments sur les symptômes peut aider à identifier des facteurs alimentaires à éviter. Un régime équilibré et varié à base de fruits, légumes, céréales complètes, protéines maigres et « bonnes » graisses est généralement recommandé.43
La tenue d’un journal alimentaire, en surveillant les effets de différents aliments sur les symptômes peut aider à identifier des facteurs alimentaires à éviter.
Objectif – une vie aussi normale que possible
L’objectif de mener vie presque normale peut être atteint par les patients ayant la FMF. La plupart d’entre eux réussissent à contrôler leurs symptômes et pratiquent des activités régulières. Il existe toujours un risque de crise. Mais des stratégies de gestion proactive réduisent l’impact de ces crises. Les patients atteints de FMF peuvent ainsi mener une vie personnelle, sociale et professionnelle épanouie. 44,45
Obtenir de l’aide
En consultant des professionnels de santé spécialistes de la FMF, comme les rhumatologues ou généticiens, les patients reçoivent les conseils d’experts et un traitement personnalisé. Le choix et le suivi d’un traitement adapté aux besoins spécifiques de chaque patient jouent un rôle important dans la prise en charge efficace des symptômes de FMF.
Par ailleurs, les relations avec les associations de patients et groupes de soutien permettent de constituer un réseau de soutien par des personnes qui comprennent toute la difficulté de vivre avec la FMF. Avec une aide adaptée, un traitement personnalisé et des perspectives encourageantes, les patients atteints de FMF parviennent à gérer leur maladie et envisagent l’avenir avec optimisme.46,47
Immunologie
Le rôle du système immunitaire
Références
1 Familial Mediterranean fever: An updated review. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5042258/
2 Familial Mediterranean Fever. American College of Rheumatology.
https://rheumatology.org/patients/familial-mediterranean-fever
3 Abdurrahman, T.; Lachmann, H.J, Familial Mediterranean fever, from pathogenesis to treatment: a contemporary review. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7672358/
4 Bhatt, H.; Cascella, M. Familial Mediterranean Fever. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/books/NBK560754/
5 Vineetha et al. Autoinflammatory syndromes: A review. Journal of Skin and Sexually Transmitted Diseases.
https://jsstd.org/autoinflammatory-syndromes-a-review/
6 Kukuy, O.L. et al. Amyloid storm: acute kidney injury and massive proteinuria, rapidly progressing to end-stage kidney disease in AA amyloidosis of familial Mediterranean fever. National Library of Medicine.
https://pubmed.ncbi.nlm.nih.gov/33291151/
7 Familial Mediterranean Fever. American College of Rheumatology.
https://rheumatology.org/patients/familial-mediterranean-fever
8Familial Mediterranean Fever. Myriad Genetics.
https://myriad.com/womens-health/diseases/familial-mediterranean-fever/
9 Amarilyo, G. Familial Mediterranean Fever. MSD Manual.
https://www.msdmanuals.com/professional/pediatrics/hereditary-periodic-fever-syndromes/familial-mediterranean-fever
10 Cush, J. Autoinflammatory Syndromes. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4489412/
11 Vineetha et al. Autoinflammatory syndromes: A review. Journal of Skin and Sexually Transmitted Diseases.
https://jsstd.org/autoinflammatory-syndromes-a-review/
12 Autoimmune Diseases. NIH, National Institute of Environmental Health Sciences.
https://www.niehs.nih.gov/health/topics/conditions/autoimmune/index.cfm
13 Vineetha et al. Autoinflammatory syndromes: A review. Journal of Skin and Sexually Transmitted Diseases.
https://jsstd.org/autoinflammatory-syndromes-a-review/
14 Familial Mediterranean Fever. NORD.
https://rarediseases.org/rare-diseases/familial-mediterranean-fever/
15 Stoffman et al. Higher than expected carrier rates for familial Mediterranean fever in various Jewish ethnic groups. NIH, National Library of Medicine.
https://pubmed.ncbi.nlm.nih.gov/10854115/
16 Familial Mediterranean Fever. NORD.
https://rarediseases.org/rare-diseases/familial-mediterranean-fever/
17 Familial Mediterranean Fever. Arthritis Foundation.
https://www.arthritis.org/diseases/familial-mediterranean-fever
18 Familial Mediterranean Fever. Arthritis Foundation.
https://www.arthritis.org/diseases/familial-mediterranean-fever
19 Amyloidosis. NHS, National Health Service.
https://www.nhs.uk/conditions/amyloidosis/
20 Infertility Causes and Pregnancy Outcome in Patients With Familial Mediterranean Fever (FMF) and Controls. The Journal of Rheumatology.
https://www.jrheum.org/content/early/2020/09/28/jrheum.200574
21 Bhatt, H.; Cascella, M. Familial Mediterranean Fever. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/books/NBK560754/
22 Ariella’s Story – Living with familial Mediterranean fever. EURORDIS.
https://www.eurordis.org/videos/ariellas-story-living-with-familial-mediterranean-fever/
23 Shohat, M. Familial Mediterranean Fever. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/books/NBK1227/
24 Meyerhoff, J.O. Familial Mediterranean Fever Workup. Medscape.
https://emedicine.medscape.com/article/330284-workup?form=fpf#c6
25 Tufan, A.; Lachmann, H.J.. Familial Mediterranean fever, from pathogenesis to treatment: a contemporary review. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7672358/
26 Familial Mediterranean Fever. Physiopedia.
https://www.physio-pedia.com/Familial_Mediterranean_Fever
27 Autosomal recessive inheritance. National Cancer Institute.
https://www.cancer.gov/publications/dictionaries/genetics-dictionary/def/autosomal-recessive-inheritance
28 Familial Mediterranean Fever. NORD.
https://rarediseases.org/rare-diseases/familial-mediterranean-fever/
29 Amarilyo, G. Familial Mediterranean Fever. MSD Manual.
https://www.msdmanuals.com/professional/pediatrics/hereditary-periodic-fever-syndromes/familial-mediterranean-fever
30 Shohat, M. Familial Mediterranean Fever. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/books/NBK1227/
31 MEFV gene. Mediline Plus.
https://medlineplus.gov/genetics/gene/mefv/#function
https://medlineplus.gov/genetics/gene/mefv/#conditions
32 MEFV gene. National Library of Medicine.
https://medlineplus.gov/genetics/gene/mefv/
33 Familial Mediterranean Fever. Arthritis Foundation.
https://www.arthritis.org/diseases/familial-mediterranean-fever
34 Familial Mediterranean Fever. Arthritis Foundation.
https://www.arthritis.org/diseases/familial-mediterranean-fever
35 Familial Mediterranean Fever. NORD.
https://rarediseases.org/rare-diseases/familial-mediterranean-fever/
36 Ozen et al. EULAR recommendations for the management of familial Mediterranean fever. BMJ Journals.
https://ard.bmj.com/content/75/4/644#block-system-main
37,38,39 Familial Mediterranean Fever. Arthritis Foundation.
https://www.arthritis.org/diseases/familial-mediterranean-fever
40 Ozen, S. et al. EULAR recommendations for the management of familial Mediterranean fever. BMJ.
https://ard.bmj.com/content/75/4/644
41 About colchicine. NHS.
https://www.nhs.uk/medicines/colchicine/about-colchicine/
42 Mansueto, P. et al. Familial Mediterranean Fever and Diet: A Narrative Review of the Scientific Literature. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9370508/
43 Familial Mediterranean Fever and Diet: A Narrative Review of the Scientific Literature. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9370508/
44 Familial Mediterranean Fever. Arthritis Foundation.
https://www.arthritis.org/diseases/familial-mediterranean-fever
45 Yüksel, C. et al. “Reality of living with familial mediterranean fever identity”: A phenomenological study.
https://cms.galenos.com.tr/Uploads/Article_30797/GMJ-61-33-En.pdf
46 Familial Mediterranean Fever. American College of Rheumatology.
https://rheumatology.org/patients/familial-mediterranean-fever
47 Familial Mediterranean Fever. NORD.
https://rarediseases.org/rare-diseases/familial-mediterranean-fever/
Févr., 2025