L’hémoglobinurie paroxystique nocturne (HPN) est une maladie hématologique qui se caractérise par un phénomène de destruction prématurée de globules rouges connu sous le nom d’hémolyse.1
L’HPN touche 950 personnes en France. C’est une maladie chronique rare qui se manifeste, entre autres, par des symptômes tels que l'anémie, la thrombose, la fatigue et le brouillard mental.2,3
Qu’est-ce que l’HPN ?
HPN est l’acronyme d’hémoglobinurie paroxystique nocturne – une maladie hématologique acquise rare, chronique, potentiellement mortelle, généralement caractérisée par un taux constamment bas d’hémoglobine, des thromboses et des symptômes invalidants. Elle survient lorsque les cellules de la moelle osseuse responsables de la fabrication des globules rouges (qui aident à transporter l’oxygène dans l’organisme) mutent et produisent des cellules sanguines défectueuses.1 Ce phénomène induit une réponse du système immunitaire qui attaque et détruit les globules rouges. La destruction prématurée des globules rouges est appelée hémolyse. Chez une personne en bonne santé, les globules rouges ont une durée de vie d'environ 120 jours. Ils sont ensuite décomposés naturellement dans l’organisme.4 Les globules rouges sont détruits de manière prématurée chez les personnes atteintes d’HPN.5
Pourquoi l’appelle-t-on hémoglobinurie paroxystique nocturne ?
Le terme HPN a été proposé au début du XXème siècle, d’après une description des symptômes observés chez les patients.
- Paroxystique désigne des symptômes d’apparition soudaine ou intermittente.
- Nocturne fait référence au moment de la survenue qui semble être la nuit (ou très tôt le matin).
- Hémoglobinurie désigne l’hémoglobine observée dans les urines (qui prennent une couleur rouge foncé ou noire).
Le terme être trompeur pour certaines personnes atteintes d’HPN, car l’affection est en réalité présente tout le temps et l’urine n’est pas systématiquement foncée.6
À quel point l’HPN est-elle rare ?
Il est difficile de déterminer exactement le nombre de personnes atteintes d’HPN, car les chiffres sont extrêmement faibles et il possible que la maladie ne soit pas diagnostiquée chez certaines personnes. D’après certaines estimations, la proportion de personnes vivant avec l’HPN se situe entre 0,5 et 2 personnes par million de la population générale, bien que des études récentes suggèrent une prévalence un peu plus élevée.7,8 Selon toute estimation, il s’agit d’une maladie très rare. L’HPN peut survenir à tout âge, mais l’âge médian au diagnostic se situe dans la trentaine.9 La fréquence est à peu près la même entre les hommes et les femmes. L’HPN est présente dans le monde entier et parmi tous les groupes ethniques.
Quels sont les symptômes de l’HPN ?
Fatigue
Jusqu’à 96 % des patients ayant une HPN ressentent de la fatigue.10
Les symptômes d’HPN peuvent varier entre les patients. Certains patients ont une forme relativement légère, tandis que d’autres peuvent présenter des symptômes sévères et invalidants.11
Les symptômes peuvent être les suivants :12,13
- Fatigue perturbant la vie quotidienne
- Brouillard mental
- Anémie
- Douleurs dorsales et abdominales
- Céphalées sévères
- Thrombose – caillots sanguins
- Hémoglobinurie – hémoglobine libre, semblable à du sang, dans les urines.
Des contractions involontaires des muscles lisses des organes internes peuvent entraîner d’autres complications telles que :
- Dyspnée – difficulté à respirer
- Dysphagie – difficulté à avaler
- Chez les hommes, dysfonction érectile.

Comment diagnostiquer l’HPN ?
Le diagnostic d’HPN peut être très tardif, car il s’agit d’une maladie très rare, peu connue des médecins. De nombreux symptômes d’HPN sont communs à plusieurs autres maladies ; en raison des symptômes très variables qu’ils sont susceptibles de présenter, les patients atteints d’HPN peuvent commencer par consulter plusieurs spécialistes (urologues, cardiologues, neurologues...) avant que le trouble sanguin et médullaire soit identifié et qu’ils puissent être orientés vers un hématologue.
Aplasie médullaire
De nombreux patients atteints d’HPN peuvent présenter simultanément un autre trouble, étroitement lié, appelé anémie aplasique acquise. Bien que le lien exact entre ces troubles n’ait pas été établi, les chercheurs pensent que l’HPN provient d’une insuffisance médullaire d’origine auto-immune, qui est la cause de la plupart des cas d’anémie aplasique acquise.14
Diagnostic initial
L’HPN peut être confirmée par une analyse spécifique du sang, la cytométrie de flux, qui identifie les cellules de l’HPN (cellules sanguines dépourvues de protéines particulières, appelées protéines à ancrage GPI) et mesure la proportion de globules rouges HPN dans le sang du patient.
Suivi diagnostique
Les médecins peuvent recommander d’autres tests de cytométrie de flux pour surveiller la progression de la maladie. Par ailleurs, deux autres analyses biologiques permettent de suivre l'évolution de la maladie :
- Lactate déshydrogénase (LDH). La LDH est une enzyme présente dans les globules rouges. Ce test donne une indication de l’importance de d’hémolyse dans l’organisme du patient.
- Numération formule sanguine (NFS). La NFS indique le nombre de globules blancs, de globules rouges et de plaquettes, ainsi que la concentration en hémoglobine. Par ailleurs, elle peut aider à identifier des anomalies de la moelle osseuse.
Quelles sont les causes de l’HPN ?
Les globules rouges, les globules blancs et les plaquettes sont produits par les cellules souches dans la moelle osseuse, par un processus appelé hématopoïèse. L’HPN résulte d’un défaut du mécanisme de production du sang – précisément, une mutation d’un gène d’une cellule souche de la moelle osseuse appelé gène PIGA (ou PIG-A).15
L’HPN est une maladie génétique acquise
L’hémoglobinurie paroxystique nocturne est causée par une mutation somatique, c’est-à-dire une modification génétique qui se produit dans une cellule, ensuite transmise aux cellules issues de la cellule mutée pendant la division cellulaire. L’HPN est donc une maladie génétique acquise. Il ne s’agit pas d’une altération génétique héréditaire, mais d’une mutation somatique du gène PIG-A.16 Cette mutation étant somatique, elle ne peut pas être transmise par les parents à leurs enfants par le biais du sperme et des ovules.17
Dans l’HPN, les cellules souches anormales produisent des copies – ou clones – d’elles-mêmes, se multipliant à chaque fois que les cellules se divisent. Les cellules anormales deviennent des globules rouges matures, porteurs d’un PIGA mutant. Ces cellules sont appelées globules rouges HPN, ou clones HPN. Les problèmes surviennent lorsque les clones HPN rencontrent le système du complément.
Hémolyse
Lorsque les globules rouges sont dégradés, l’hémoglobine qu'ils contiennent est libérée. L’hémoglobine est la partie rouge des globules rouges qui transporte l’oxygène dans l’organisme. La libération d’hémoglobine dans le plasma est à l’origine de nombreux symptômes de l’HPN.
On distingue deux types, ou mécanismes, d’hémolyse : l’hémolyse intravasculaire (HIV) et l’hémolyse extravasculaire (HEV). Tous deux se produisent après le début d’une cascade d’activité dans le système du complément, qui fait partie du système immunitaire.18
- HIV – les clones HPN sont détruits dans les vaisseaux sanguins.
- HEV – les clones HPN sont détruits dans le foie et la rate.
Le système du complément
Le système, ou cascade, du complément est un groupe d’environ 60 protéines présentes dans le sang. Ces protéines sont appelées ainsi car elles complètent le travail des globules blancs dans la lutte contre les infections Elles conservent en permanence un faible niveau d’activité. Si des cellules anormales – des bactéries, des virus ou d’autres pathogènes – sont détectées dans l’organisme, ces protéines deviennent plus actives, et attaquent et détruisent les cellules anormales.
Il existe deux protéines essentielles pour l’HPN dans la cascade du complément, appelées C3 et C5. Ces dernières sont successivement activées lorsque le système immunitaire détecte des agents pathogènes.
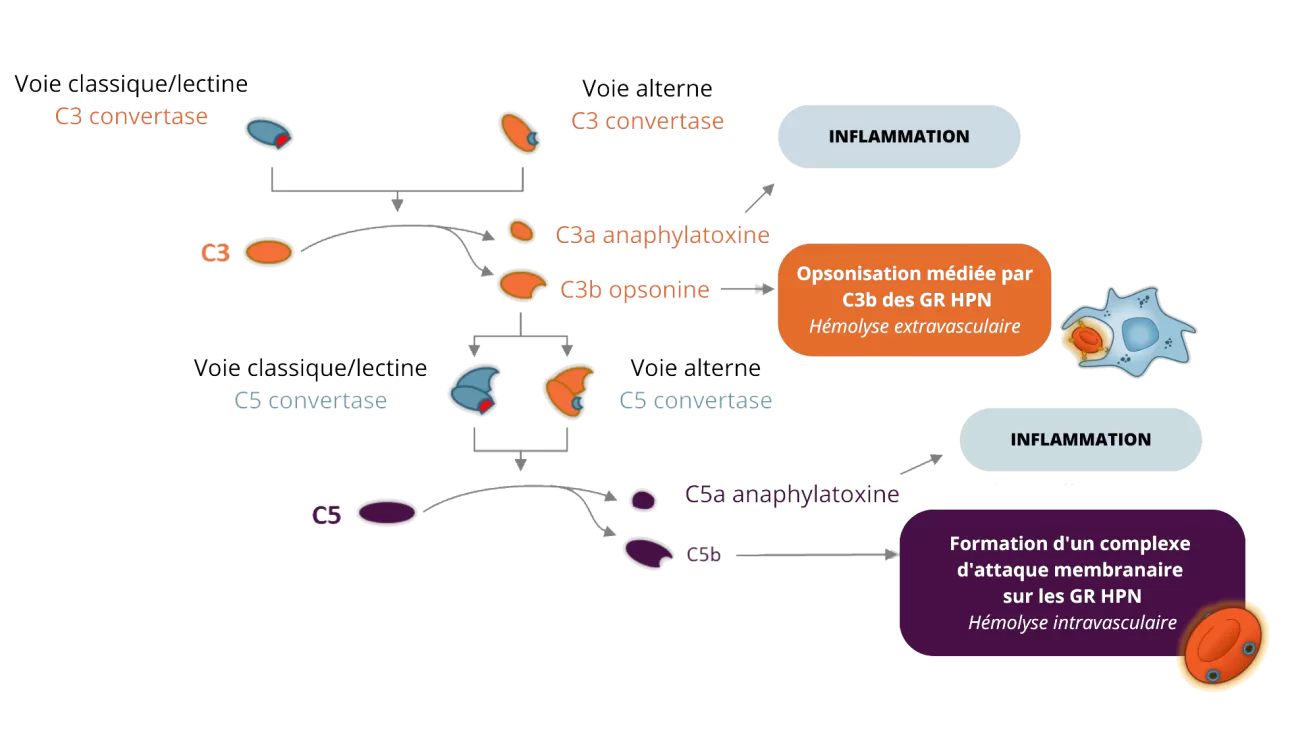
En réponse à la détection d’un agent pathogène (ou d’une menace perçue comme un pathogène) :
- La protéine C3 est activée et scindée (clivée) en C3a et C3b
- La protéine C3b active C5
- La protéine C5 est clivée en C5a et C5b
- Le complexe d’attaque membranaire (CAM) est formé, agissant au sein du système du complément pour attaquer et détruire les agents pathogènes.
Les globules rouges normaux portent un bouclier de protéines protectrices, appelées protéines à ancrage GPI, dont la fonction est d’empêcher le système du complément de les attaquer. Le gène qui fabrique ce bouclier protecteur est le gène PIG-A.
Les clones HPN, produits par les cellules souches porteuses du gène PIG-A déficient sont dépourvus de ce bouclier protecteur. Par conséquent, les protéines du système du complément confondent les clones HPN avec des agents pathogènes, et les attaquent et les détruisent. Il s’agit d’une réponse auto-immune, ce qui signifie que le système de défense naturel de l’organisme ne parvient pas à distinguer ses propres cellules de cellules étrangères – entraînant une destruction prématurée ou excessive des globules rouges.
Quels sont les traitements disponibles ?
La greffe de moelle osseuse est le seul traitement à visée curative possible. En cas de réussite, cette procédure peut guérir définitivement le patient atteint d’HPN. Toutefois, la greffe de moelle osseuse comporte des risques qui l’emportent souvent sur les bénéfices attendus, et n’est donc pas envisageable.
Ainsi, en l’absence de traitement curatif, ces approches peuvent limiter l’impact de la maladie sur la vie des patients.
L'introduction des inhibiteurs du complément a grandement amélioré la prise en charge, avec un allongement de l’espérance de vie des patients atteints d’HPN.24
Comment vivre mieux avec l’HPN ?
Comprendre les aspects scientifiques, les symptômes et le traitement de l’HPN peut aider les patients atteints d’HPN à mieux appréhender leur santé et leurs soins. Ils peuvent devenir acteurs de leur santé en connaissant mieux leurs symptômes et en expliquant leurs objectifs concernant l’HPN à l’équipe soignante.
Les limites imposées par les symptômes et le traitement de l’HPN amènent souvent les patients à renoncer à certaines possibilités offertes par la vie.25,26,27,28 Les consultations avec leur équipe soignante peuvent être une bonne occasion de discuter de leurs aspirations au quotidien et des objectifs pour l’avenir.
Avec un réseau de soutien, les patients peuvent atteindre ces objectifs. Il existe plusieurs possibilités de soutien : famille ou amis, associations de patients ou encore ressources externes.
Les échanges avec le médecin traitant ou l’engagement au sein d'une association de patients peuvent apporter un soutien et des perspectives aux patients en attente de changement.
Hématologie
Spécialité médicale étudiant le sang et ses maladies
Références
1 Hill A, DeZern AE, Kinoshita T & Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers 2017;3:17028.Diagnosis and management of paroxysmal nocturnal hemoglobinuria Charles Parker et al
https://ashpublications.org/blood/article/106/12/3699/109767/Diagnosis-and-management-of-paroxysmal-nocturnal
2 hemoglobinuria Charles Parker et al
https://ashpublications.org/blood/article/106/12/3699/109767/Diagnosis-and-management-of-paroxysmal-nocturnal
3 Peffault de Latour R, Sicre de Fontbrune F, Leblanc T, Brindel I. Livret d’information patient - FOIRE AUX QUESTIONS - PRISE EN CHARGE D’UNE HÉMOGLOBINURIE PAROXYSTIQUE NOCTURNE. Centre de Référence des aplasies médullaires, editor. 2020.
4 Frontiers in Physiology: How Do Red Blood Cells Die?
https://www.frontiersin.org/articles/10.3389/fphys.2021.655393/full
5 National Center for Biotechnology Information: Paroxysmal nocturnal hemoglobinuria (PNH), MedGen UID: 7471
https://www.ncbi.nlm.nih.gov/medgen/7471
6 PNH National Service, Leeds & London.
https://www.pnhleeds.co.uk/patients/what-is-pnh/
7 https://medlineplus.gov/genetics/condition/paroxysmal-nocturnal-hemoglobinuria/#frequency
8 https://pubmed.ncbi.nlm.nih.gov/34060690/
9 Paroxysmal Nocturnal Hemoglobinuria, NORD
https://rarediseases.org/rare-diseases/paroxysmal-nocturnal-hemoglobinuria/
10 Lima M. Laboratory studies for paroxysmal nocturnal hemoglobinuria, with emphasis on flow cytometry. Pract Lab Med 2020;20.
11 Lima M. Laboratory studies for paroxysmal nocturnal hemoglobinuria, with emphasis on flow cytometry. Pract Lab Med 2020;20.
12 Meyers G, Weitz I, Lamy T, et al. Disease-Related Symptoms Reported across a Broad Population of Patients with Paroxysmal Nocturnal Hemoglobinuria. Blood 2007;110:3683.
13 Mitchell R, Salkeld E, Chisolm S, et al. Path to Diagnosis of Paroxysmal Nocturnal Hemoglobinuria: The Results of an Exploratory Study Conducted by the Aplastic Anemia and MDS International Foundation and the National Organization for Rare Disorders Utilizing an Internet-Based Survey. SM Clin Med Oncol 2017;1:1001.
14 Paroxysmal Nocturnal Hemoglobinuria, NORD
https://rarediseases.org/rare-diseases/paroxysmal-nocturnal-hemoglobinuria/
15 National Center for Biotechnology Information: Paroxysmal nocturnal hemoglobinuria (PNH), MedGen UID: 7471
https://www.ncbi.nlm.nih.gov/medgen/7471
16 https://pubmed.ncbi.nlm.nih.gov/8306954/
17 https://www.cancer.gov/publications/dictionaries/cancer-terms/def/somatic-mutation
18 Devos T, Meers S, Boeckx N et al., Diagnosis and management of PNH: Review and recommendations from a Belgian expert panel. Eur J Haematol 2018;101:737-749.
19 Hill A, DeZern AE, Kinoshita T & Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers 2017;3:17028.
20 Cho H. Complement regulation: physiology and disease relevance. Korean J Pediatr 2015;58:239–44.
21 Merle NS, Church SE, Fremeaux-Bacchi V & Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol 2015;6:262.
22 Hill et al. Nat Rev Dis Primers
23 Risitano et al. Front Immunol 2019
24 M Loschi et al. Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no-treatment study
https://pubmed.ncbi.nlm.nih.gov/26689746/
25 Dingli D, Matos JE, Lebrhaunt K, et al. Clinical Burden of Paroxysmal Nocturnal Hemoglobinuria Among Patients Receiving C5 Inhibitors in the United States. Blood. 2020;136(Supplement 1).
26 Dingli D, Matos JE, Lebrhaunt K, et al. Work Productivity Loss and Quality of Life in Paroxysmal Nocturnal Hemoglobinuria Among Patients Receiving C5 Inhibitors in the United States. 2020;136(Supplement 1):3.
27 Dingli D, Matos JE, Lehrhaupt K, et al. The burden of illness in patients with paroxysmal nocturnal hemoglobinuria receiving treatment with the C5-inhibitors eculizumab or ravulizumab: results from a US patient survey. Ann Hematol. 2022;101(2):251–263.
28 Bektas et al. Paroxysmal nocturnal hemoglobinuria: patient journey and burden of disease. Manag Care Spec Pharm. 2020;26:S8-S14.
Févr., 2025