L’hémophilie est un trouble de la coagulation sanguine. Il existe deux types d’hémophilie : l’hémophilie A et l’hémophilie B, plus rare. Ces deux types d’hémophilie n’ont pas la même cause, mais leurs symptômes sont identiques.1 L’hémophilie est bien plus fréquente, et généralement plus sévère, chez l’homme que chez la femme.2,3 La maladie se caractérise par la formation lente de caillots sanguins insuffisants, à l’origine de saignements prolongés et de saignements internes spontanés.
Qu’est-ce que l’hémophilie ?
Chez une personne non hémophile, un traumatisme – interne ou externe – entraînant un saignement déclenche un processus appelé cascade de la coagulation. Cette cascade correspond à une série de réactions dans la circulation sanguine, aboutissant à l’hémostase – l’arrêt du saignement.
En réalité, cette cascade débute par deux séries d'étapes d’origine distincte, appelées voie intrinsèque et voie extrinsèque. Ces deux voies convergent vers une troisième voie, la voie commune. Le mécanisme de coagulation sanguine fait appel à plusieurs facteurs de coagulation circulant dans le sang. Chaque facteur active le suivant, dans un ordre précis.4 Si une étape de ce processus complexe échoue, il est possible que le sang ne coagule pas correctement, et que des saignements excessifs apparaissent. Lors d’une hémorragie, les différents facteurs de coagulation interviennent pour former un caillot sanguin, colmater la brèche et arrêter le saignement.
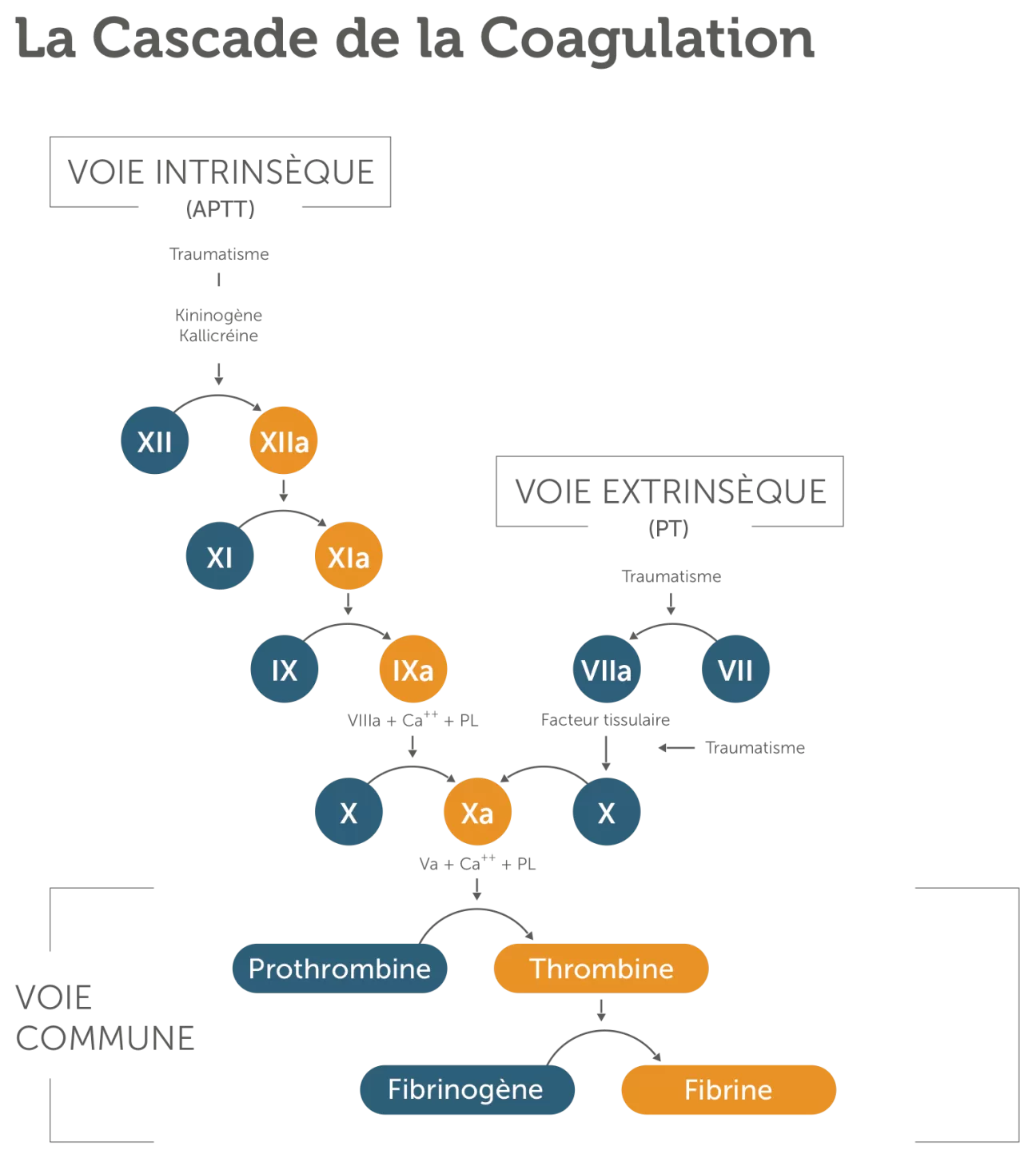
Les facteurs de coagulation VIII et IX sont des protéines du sang impliquées dans le processus de coagulation.
L'hémophilie A est due à un déficit en facteur VIII.
L'hémophilie B est causée par un déficit en facteur IX. 6
Chez les personnes atteintes d'hémophilie, l'un de ces facteurs vient à manquer ou n’est pas suffisamment efficace. Le caillot se forme alors plus lentement et leur sang coule plus longtemps après une blessure.
Des saignements peuvent également survenir dans les articulations et les muscles sans traumatisme apparent. On parle alors de saignements spontanés. 5
Y a-t-il d’autres maladies hémorragiques similaires à l’hémophilie A ou B ?
Il existe d’autres maladies hémorragiques rares :
L’hémophilie C, aussi appelée maladie de Rosenthal. Il s’agit d’une forme légère d’hémophilie, touchant les deux sexes, causée par un déficit en facteur XI.7 Bien plus rare que l’hémophilie A ou B, elle touche environ 1 personne sur un million.8
La maladie de von Willebrand, une maladie hémorragique génétique, caractérisée par un déficit en, ou une anomalie du facteur von Willebrand (vWF), une protéine du sang importante dans la formation des caillots.9
Dans de très rares cas, il est possible de naître avec, ou de développer les déficits suivants : déficit en facteur I (fibrinogène), déficit en facteur II (prothrombine), déficit en facteur V, déficit en facteur VII, déficit en facteur X, déficit en facteur XII ou déficit en facteur XIII.10
La thrombopénie immune ou purpura thrombopénique immunologique (PTI), une maladie auto-immune rare, affectant la production des plaquettes. Le PTI est différent de l’hémophilie, bien que certains symptômes soient similaires. Les patients atteints de PTI ont un taux réduit de plaquettes, mais le mécanisme de coagulation fonctionne par ailleurs normalement.11,12
Quelle est la fréquence de l’hémophilie ?13
L’hémophilie A touche environ un 1 garçon sur 5000 à la naissance. L’hémophilie B est plus rare. Elle touche environ 1 garçon sur 30 000. Les formes sévères d’hémophilie sont très rares chez la femme. Environ 30 % à 50 % des femmes porteuses peuvent présenter une forme légère d’hémophilie. L’hémophilie touche toute la population, quelle que soit l’origine ethnique, partout dans le monde.14
Quels sont les symptômes de l’hémophilie ?15
Les symptômes de l’hémophilie sont les saignements. Ils peuvent se produire n’importe où dans le corps mais ont le plus souvent lieu dans les articulations ou dans les muscles.
- Ecchymoses (« bleus »)
- Saignements musculaires (hématomes) et articulaires (hémarthroses), pouvant causer :
o Endolorissement
o Gonflement
o Douleur et raideur
o Difficulté à mobiliser une articulation ou un muscle - Saignements spontanés : hémorragie interne soudaine, sans cause apparente, susceptible d’engager le pronostic vital
- Saignement prolongé après une intervention chirurgicale ou une extraction dentaire
- Saignement prolongé après un accident, en particulier après un traumatisme crânien16
Comment diagnostiquer l’hémophilie ?
Il peut être recommandé d'établir un diagnostic de certitude chez les personnes montrant certains ou tous les symptômes d’hémophilie, en particulier en présence d’antécédents familiaux d’hémophilie.17,18 En première intention, il peut être proposé au patient de réaliser un bilan d’hémostase à partir d’un prélèvement sanguin. En cas de suspicion d’hémophilie, le patient peut ensuite être orienté vers un centre de traitement de l’hémophilie afin de mesurer l’activité du facteur VIII et du facteur IX.
Niveaux de sévérité de l’hémophilie et signification19,20
Le niveau de sévérité de l'hémophilie est lié à la quantité de facteur de coagulation présent dans le sang.
Pas d’hémophilie
Pourcentage de l’activité normale du facteur dans le sang
40-150%
Hémophilie mineure
Le taux de facteur est compris entre 5 et 40 % du taux normal.
Survenue du saignement
Après un traumatisme ou une intervention chirurgicale
Hémophilie modérée
Le taux de facteur est compris entre 1 et 5 % du taux normal.
Survenue du saignement
Suite à un traumatisme mineur ou parfois spontané
Hémophilie sévère
Le taux de facteur dans le sang est inférieur à 1 % du taux normal.
Survenue du saignement
Saignements spontanés dans les articulations et les muscles
Moins il y a de facteur de coagulation dans le sang, plus l’hémophilie est sévère, et plus les saignements sont graves et spontanés.
Quelle sont les causes de l’hémophilie ?21
L’hémophilie survient en réponse à une modification, ou mutation, du gène responsable de la production du facteur de coagulation VIII ou IX, aboutissant à une production insuffisante du facteur.
Dans l’hémophilie A, le gène responsable de la production du facteur VIII, appelé gène F8, est altéré.
Dans l’hémophilie B, le gène responsable de la production du facteur IX, appelé gène F9, est altéré.
En règle générale, l’hémophilie est une maladie héréditaire, transmise par les parents à leurs enfants. Toutefois, dans environ un tiers des cas d’hémophilie A ou B, il n’y a pas d’antécédents familiaux de la maladie. L’hémophilie héréditaire peut en effet provenir d’une mutation germinale spontanée – c’est-à-dire en présence d’une modification de l’ADN des enfants, absente de l’ADN parental mais pouvant être transmise à la génération suivante.22,23 Ce type de modification génique est également appelé mutation de novo.24,25

Au 19ème siècle, la Reine d'Angleterre Victoria, porteuse du gène de l'hémophilie B, la transmit à son plus jeune fils, Léopold, ainsi qu'à deux de ses filles. En tout, plus de 20 de ses descendants furent atteints d'hémophilie. Depuis, l'hémophilie est également appelée "maladie royale".
Hémophilie acquise
Dans de rares cas, une personne peut devenir hémophile au cours de sa vie, même en l’absence d’antécédents familiaux et de mutation spontanée. Cette forme d’hémophilie est dite acquise (HA). L’HA est une maladie auto-immune dans laquelle l’organisme attaque par erreur des cellules ou tissus sains. Dans l’HA, l’organisme produit des anticorps, appelés inhibiteurs, qui attaquent des facteurs de coagulation, le facteur VIII le plus souvent. L’HA est principalement observée chez les personnes âgées. On estime qu’elle touche environ une personne sur un million.26
Comment se transmet l’hémophilie ?
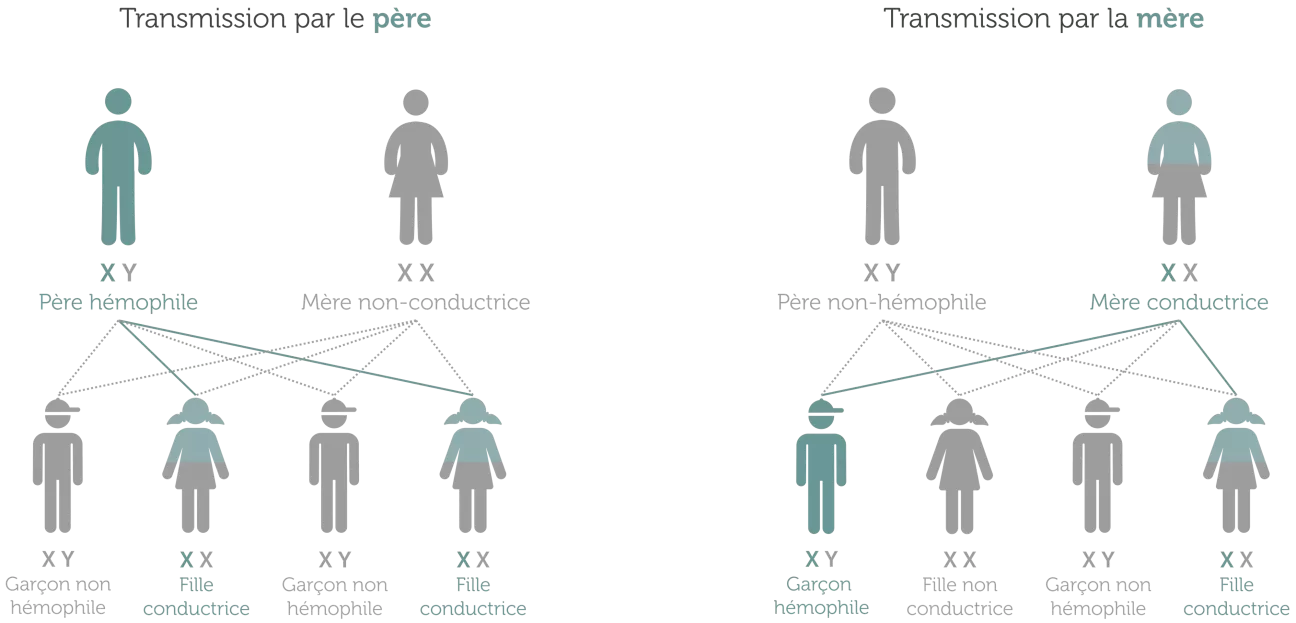
Les hémophilies A et B sont des maladies à transmission récessive, liée à l’X. Quelques notions de génétique sont nécessaires pour comprendre ce que cela signifie.
Chaque cellule du corps humain contient normalement 23 paires de chromosomes. Ces paires de chromosomes contiennent tout notre matériel génétique. Chaque chromosome porte plusieurs centaines ou milliers de gènes. Les gènes contiennent les instructions nécessaires à la fabrication de protéines. 22 des paires de chromosomes, appelés autosomes, sont identiques chez l’homme et la femme. La 23ème paire, celle des chromosomes sexuels, est différente entre l’homme et la femme. Les femmes possèdent deux copies du chromosome X (XX), tandis que les hommes ont un chromosome X et un chromosome Y (XY).27,28
Le gène F8 et le gène F9, respectivement responsables de la production des facteurs VIII et IX, sont situés sur le chromosome X. Leurs conséquences sont donc différentes chez l’homme et la femme.
Si un femme présente une mutation récessive responsable de l’hémophilie (A ou B) sur un seul de ses chromosomes X, elle sera conductrice. Cela signifie qu’elle ne développera pas la maladie, ou peut-être une forme atténuée, mais qu’elle peut transmettre la mutation à ses enfants. En moyenne, la moitié de ses fils seront hémophiles et la moitié de ses filles seront conductrices de la mutation.
Si un homme présente la même mutation sur le chromosome X, il développera la maladie car il ne possède pas d’autre chromosome X sans mutation. Il transmettra le gène muté à toutes ses filles par l’intermédiaire du chromosome X qu'il apporte. Il ne peut pas le transmettre à ses fils, car il ne leur apporte uniquement le chromosome Y.
Quels sont les traitements disponibles ?
Par le passé, les transfusions à l’hôpital étaient un des rares traitements disponibles pour l’hémophilie et l’espérance de vie des patients ayant une forme sévère était réduite. Des progrès importants ont été réalisés ces dernières décennies et années.
Les traitements actuels ont révolutionné la prise en charge des patients hémophiles, en leur permettant de repousser les limites conventionnelles et de mener une vie plus active. Grâce aux traitements modernes, les hémophiles peuvent non seulement avoir plus d’activités physiques, notamment sportives, mais aussi vivre au quotidien sans redouter en permanence les épisodes de saignements. Pour les enfants, ces progrès signifient la possibilité de participer à différentes activités avec leurs camarades et rendent leur vie plus inclusive.
Types de traitements de l’hémophilie
Médicaments dérivés du plasma
Les concentrés de facteurs sont dérivés du plasma humain. Ces produits, issus de la purification de dons de plasma sanguin, constituent une source naturelle de facteurs de coagulation. Des protocoles rigoureux de dépistage et de purification sont appliqués pour minimiser le risque de transmission d'infections. Les facteurs dérivés du plasma ont une demi-vie proche de la demi-vie physiologique des facteurs de coagulation.29
Médicaments recombinants
Les concentrés de facteurs recombinants sont produits par des techniques de génie génétique. Les facteurs de coagulation recombinants sont développés en laboratoire. Cette méthode permet de garantir l’homogénéité et la pureté du produit, réduisant ainsi le risque de contamination et d’effets indésirables. Les concentrés recombinants peuvent être des facteurs à demi-vie standard, prolongée ou très allongée.30
Facteur à demi-vie standard ou Standard Half-Life factor (SHL)
Les traitements par concentrés de facteurs standards comportent des injections de facteurs de coagulation à demi-vie classique, nécessitant des administrations régulières pour maintenir des taux suffisants dans le sang.31 Cette forme de traitement peut être utilisée à la demande ou en prophylaxie.32
Facteur à demi-vie prolongée ou Extended Half-Life Factor (EHL)
Les concentrés de facteurs à demi-vie prolongée sont conçus pour rester actifs plus longtemps dans le sang. Ils permettent d’obtenir des taux plus élevés de facteurs avec des injections moins fréquentes. Cette approche vise à améliorer le contrôle des saignements tout en réduisant la fréquence du traitement, en particulier pour les schémas prophylactiques. 33,34,35, 36.
Traitement non substitutif
Ces thérapies ne visent pas à remplacer le facteur de coagulation manquant, mais consistent à mimer l’action du facteur de coagulation manquant ou à limiter l’action de facteurs ayant une action opposée au facteur manquant (anticoagulants) pour rétablir une coagulation normale.35
Thérapie génique
La thérapie génique consiste en l’introduction d'une copie (un allèle) fonctionnelle du gène codant pour le facteur de coagulation dans les cellules du patient. Elle permet à l’organisme de produire le facteur de coagulation déficient. Cette approche vise à corriger à long terme la cause génétique sous-jacente de l’hémophilie. Aucune thérapie génique n'est commercialisée en France à ce jour.36
Développement d’anticorps
Les traitements substitutifs sont efficaces pour la plupart des patients. Toutefois, dans certains cas, l’organisme peut réagir au facteur de coagulation qu’il considère comme une substance étrangère. Le système immunitaire développe alors des anticorps appelés inhibiteurs, qui rendent le facteur de coagulation injecté moins efficace, voire totalement inefficace. Les anticorps peuvent disparaître spontanément après une période de traitement régulier. L’injection de doses plus élevées peut aussi être nécessaire pendant un certain temps. Les anticorps peuvent parfois persister dans l’organisme. Les saignements doivent dans ce cas être contrôlés par un autre traitement.37
Liberate Life – "Une vie au-delà de l’hémophilie"
Liberate Life est notre engagement à long terme envers tous ceux qui vivent avec l’hémophilie, leurs familles et leurs amis, et les professionnels de santé qui prennent soin d’eux.
Sobi s’engage dans la sensibilisation des patients et l’accompagnement des soignants pour le suivi, la prévention et la prise en charge des dommages articulaires et des douleurs associées.
Engagement à long terme
Notre engagement nous pousse à toujours innover en matière de traitements de l’hémophilie dans le seul but de mettre à disposition des personnes hémophiles de nouvelles options pouvant les aider à vivre la vie qu’ils aimeraient avoir.
Notre savoir-faire
Nos compétences dans le domaine de l’hémophilie sont une plateforme pour l’avenir.
L’héritage d’innovation de Sobi dans l’hémophilie est un engagement à long terme vis-à-vis des patients et de leurs soignants et une base solide sur laquelle nous nous appuyons pour continuer à être pionniers dans nos recherches.
Hématologie
L'hématologie est une spécialité médicale traitant les maladies du sang
Références
1Understanding Haemophilia, The Haemophilia Society
https://haemophilia.org.uk/wp-content/uploads/2017/04/Understanding_haemophilia_WEB.pdf
2Genetic causes of haemophilia in women and girls. Table 1. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8132474/
3Haemophilia. Better Health Channel.
https://www.betterhealth.vic.gov.au/health/conditionsandtreatments/haemophilia
4Chaudhry, R.; Usama, S.M.; Babiker, H.M. Physiology, Coagulation Pathways. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/books/NBK482253/
5Haemophilia. The Haemophilia Society.
https://haemophilia.org.uk/bleeding-disorders/haemophilia-a-and-b/
6Haemophilia, The Haemophilia Society.
https://haemophilia.org.uk/bleeding-disorders/haemophilia-a-and-b/
7Factor XI Deficiency. Medscape.
https://emedicine.medscape.com/article/209984-overview
8Thejus, J. et al. Hemophilia C: A Case Report With Updates on Diagnosis and Management of a Rare Bleeding Disorder. National Library of Medicine.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7153668/
9What is von Willebrand Disease (VWD)? bleedingdisorders.com.
https://www.bleedingdisorders.com/about/von-willebrand-disease
10Other rare bleeding disorders. bleedingdisorders.com.
https://www.bleedingdisorders.com/about/types-of-bleeding-disorders
11Hemophilia, Differential Diagnosis. Rare Disease Advisor.
https://www.rarediseaseadvisor.com/disease-info-pages/hemophilia-differential-diagnosis/
12Difference Between Thrombocytopenia and Hemophilia. Difference Between.com.
https://www.differencebetween.com/difference-between-thrombocytopenia-and-vs-hemophilia/
13Understanding Haemophilia, The Haemophilia Society.
https://haemophilia.org.uk/wp-content/uploads/2017/04/Understanding_haemophilia_WEB.pdf #3
14Understanding Haemophilia, The Haemophilia Society.
https://haemophilia.org.uk/wp-content/uploads/2017/04/Understanding_haemophilia_WEB.pdf #3
15E-Learning. World Federation of Haemophilia.
https://elearning.wfh.org/elearning-centres/hemophilia/
16E-Learning. World Federation of Haemophilia.
https://elearning.wfh.org/elearning-centres/hemophilia/
17Hemophilia. Medline Plus.
https://medlineplus.gov/hemophilia.html
18Hemophilia. Cleveland Clinic.
https://my.clevelandclinic.org/health/diseases/14083-hemophilia
19Understanding Haemophilia, The Haemophilia Society.
https://haemophilia.org.uk/wp-content/uploads/2017/04/Understanding_haemophilia_WEB.pdf #2
20About Haemophilia. NHS Southern Haemophilia Network.
https://www.southernhaemophilianetwork.org/about/haemophilia/what-is/
21How Hemophilia is Inherited. CDC. Centers for Disease Control and Prevention.
https://www.cdc.gov/ncbddd/hemophilia/inheritance-pattern.html
22Hemophilia A, National Hemophilia Foundation.
https://www.hemophilia.org/bleeding-disorders-a-z/types/hemophilia-a
23Hemophilia B, National Hemophilia Foundation.
https://www.hemophilia.org/bleeding-disorders-a-z/types/hemophilia-b
24Hemophilia A. NORD.
https://rarediseases.org/rare-diseases/hemophilia-a/
25Haemophilia B. ScienceDirect.
https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/haemophilia-b
26Acquired Hemophilia. NORD.
https://rarediseases.org/rare-diseases/acquired-hemophilia/
27How many chromosomes do people have? Medline Plus.
https://medlineplus.gov/genetics/understanding/basics/howmanychromosomes/
28Human Genome Project FAQ, NIH.
https://www.genome.gov/human-genome-project/Completion-FAQ
29Treatment of Haemophilia. Centers for Disease Control and Prevention.
https://www.cdc.gov/ncbddd/hemophilia/treatment.html
30Treatment of Haemophilia. Centers for Disease Control and Prevention.
https://www.cdc.gov/ncbddd/hemophilia/treatment.html
31Haemophilia Treatment. Irish Haemophilia Society.
https://haemophilia.ie/about-bleeding-disorders/haemophilia/haemophilia-treatment-2/
32SHL/EHL Fact Sheet. World Federation of Hemophilia.
https://www1.wfh.org/publications/files/pdf-2365.pdf
33Berntorp E, Negrier C, Gozzi P, Blaas PM, Lethagen S. Dosing regimens, FVIII levels and estimated haemostatic protection with special focus on rFVIIIFc. Haemophilia. 2016;22(3):389-396.
34Current Treatments. National Bleeding Disorders Foundation.
https://www.hemophilia.org/bleeding-disorders-a-z/treatment/current-treatments
35Shapiro et al. Real-world data demonstrate improved bleed control and extended dosing intervals for patients with haemophilia B after switching to recombinant factor IX Fc fusion protein (rFIXFc) for up to 5 years. Haemophilia 2020 Nov;26(6):975-983
36Tagliaferri A, Matichecchia A, Rivolta GF, Riccardi F, Quintavalle G, Benegiamo A, Rossi R, Coppola A. Optimising prophylaxis outcomes and costs in haemophilia patients switching to recombinant FVIII-Fc: a single-centre real-world experience. Blood Transfus. 2020;18(5):374-385
37Current Treatments. National Bleeding Disorders Foundation.
https://www.hemophilia.org/bleeding-disorders-a-z/treatment/current-treatments
38Inhibitors. The Haemophilia Society.
https://haemophilia.org.uk/bleeding-disorders/inhibitors/
Févr., 2025