L’Hémoglobinurie Paroxystique Nocturne, ou maladie de Marchiafava-Micheli, a été décrite pour la première fois à la fin du XIXème siècle.1 Le terme HPN a été choisie sur la base d'une description de la triade de symptômes observés chez les patients :
• Hémoglobinurie, caractérisant la présence d'hémoglobine libérée par la destruction des globules rouges dans les urines.2
• Paroxystique, pour les poussées soudaines2
• Nocturne, c’est-à-dire survenant la nuit2
L’HPN c’est quoi ?
L’hémoglobinurie paroxystique nocturne (HPN) est une maladie hématologique qui se caractérise par un phénomène de destruction prématurée de globules rouges connu sous le nom d’hémolyse.1
L’HPN touche 600 personnes en France. C’est une maladie chronique rare dont la présentation clinique et la gravité varient, pouvant retarder ou compliquer son diagnostic. L’errance diagnostic pour ces patients peut aller jusqu’à 5 ans. Potentiellement mortelle, les personnes atteintes d’HPN non diagnostiquées et non traitées risquent de mourir d’une thrombose, d’une hémorragie ou d’une infection. En effet, en l'absence de traitement 20 à 40% des patients atteints d'HPN décèdent dans les 5 à 6 ans suivant le diagnostic. 3,4,5,6
Quelle est l’origine de la maladie ?
L’hémoglobinurie paroxystique nocturne survient lorsque les cellules souches hématopoïétiques (CSH) responsables de la fabrication des cellules sanguines de la moelle osseuse, mutent et commencent à produire des cellules sanguines défectueuses ayant perdues l’expression à leur surface de certaines protéines. 1,2,7
La maladie résulte d'un défaut dans le mécanisme de production du sang - plus précisément une mutation dans un gène d'une CSH appelé le gène PIGA. Ces cellules souches porteuses de mutations du gènes PIG-A vont se multiplier et conduire à la formation de cellules sanguines matures porteuses de la même mutation. Les CSH mutantes forment alors une population de cellules sanguines mutantes appelées clones HPN. 1,8,9
Les manifestations cliniques d’HPN surviennent lorsqu’un clone de CSH porteur de mutations somatiques du gène PIGA acquiert un avantage de croissance et se différencie, créant une proportion importante de cellules sanguines matures déficitaires en protéines GPI-ancrées. Les globules rouges et granulocytes déficitaires en CD55 et CD59 qui en résultent sont appelés collectivement clones HPN. Le pourcentage de globules rouges ou granulocytes présentant un déficit en CD55 et CD59 est appelé taille du clone HPN. 1,10

Dans l’HPN, les mutations du gène PIGA entraînent la perte de CD55 et CD59, qui provoque à son tour la destruction des globules rouges médiée par le complément.1
Protéines GPI-ancrées : CD55 et CD59
Le gène PIGA est requis pour la synthèse des ancres GPI, et donc pour la présence de protéines spécialisées à la surface des cellules1 CD55 et CD59 sont des types de protéines GPI-ancrées. Ce sont des régulateurs négatifs du complément qui empêchent les cellules de l’organisme d’être attaquées par le système du complément.1

Le système du complément est une composante centrale du système immunitaire impliquée dans la défense contre les agents pathogènes, l’homéostase de l’hôte et cellulaire, et la régulation de la réponse immunitaire. Composé de protéines, de convertases, de récepteurs et de régulateurs, le système du complément facilite l’élimination des agents pathogènes nocifs et des cellules de l’organisme qui sont endommagées par des phénomènes d’inflammation, de lyse directe, d’opsonisation et de phagocytose. 11,12,13,14

Dans l’HPN, les globules rouges HPN vont être reconnus par le système du complément comme des organismes étrangers en raison de leur modification génétique. L’hémolyse des globules rouges HPN mutant médiée par le système du complément libère dans le plasma l’hémoglobine qu’ils contenaient. Cette libération provoque une anémie modérée à sévère et un ensemble de symptômes et de complications (élévations des taux hémoglobine, thrombose, libération de LDH et de bilirubine). 1,15,16
Il existe deux types hémolyses :
- Hémolyse intra-vasculaire : conséquence de la formation du complexe d’attaque membranaire sur les globules rouges HPN à l’intérieur des vaisseaux sanguins
- Hémolyse extra-vasculaire : résultant de l’opsonisation des globules rouges HPN médiée par C3b qui seront détruits dans le foie et la rate
Quels impacts pour le patient ?
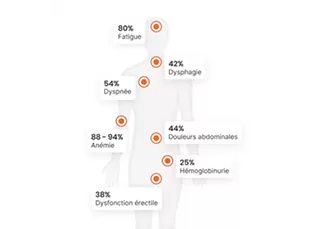
L’HPN est responsable de nombreux signes et symptômes, qui peuvent compliquer sérieusement le quotidien des patients.
De multiples manifestations cliniques chroniques et invalidantes sont souvent présentes dans l’HPN, il peut s’agir notamment de taux d’hémoglobine constamment bas, d’anémie, de fatigue, d’essoufflement, de difficulté à avaler, de maux de tête, d’urine foncée, de douleurs abdominales ou encore de dysfonction érectile.4,5
Références
- Hill, A., DeZern, A., Kinoshita, T. et al. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers 3, 17028 (2017). https://doi.org/10.1038/nrdp.2017.28
- Livret d’information HPN https://marih.fr/wpcontent/uploads/2019/12/livret_hpn_digitaljuillet_2020.pdf
- Panse J, Sicre de Fontbrune F, Burmester P, Piggin M, Matos JE, Costantino H, Wilson K, Hakimi Z, Nazir J, Desgraz R, Fishman J, Persson E, Kulasekararaj A. The burden of illness of patients with paroxysmal nocturnal haemoglobinuria receiving C5 inhibitors in France, Germany and the United Kingdom: Patient-reported insights on symptoms and quality of life. Eur J Haematol. 2022 Oct;109(4):351-363.
- Sicre de Fontbrune F, Burmester P, Piggin M, Matos JE, Costantino H, Wilson K, Hakimi Z, Nazir J, Desgraz R, Fishman J, Persson E, Panse J. The burden of illness of patients with
- Röth A, et al. Screening and Diagnostic Clinical Algorithm for Paroxysmal Nocturnal Hemoglobinuria: Expert Consensus. Eur J Haematol. 2018 Jul;101(1):3-11.
- Petropoulou AD, et al. ECLIPSE: a French Study Concerning the Diagnosis of Paroxysmal Nocturnal Hemoglobinuria (PNH). Blood. 2010;116(21):5134.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13(1):110-117. doi:10.1002/j.1460-2075.1994.tb06240.x
- Parker et al. Blood 2005
- Brodsky Expert Rev Hematol 2009.
- Pu et al. Eur J Haematol 2011.
- Merle et al. Front Immunol 2015a
- Janeway et al. 2001.
- Merle et al. Front Immunol 2015b
- Dunkelberger & Song Cell Res 2010
- Kanakura et al. 2017
- Hill et al. Blood 2013
- Debureaux PE, et al. Categorizing hematological response to eculizumab in paroxysmal nocturnal hemoglobinuria: a multicenter real-life study. Bone Marrow Transplant. 2021 Oct;56(10):2600-2602.
- Risitano AM and Marotta S. Toward complement inhibition 2.0: Next generation anticomplement agents for paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2018 Aug;93(4):564-577.
- Berentsen S, et al. Novel insights into the treatment of complement-mediated hemolytic anemias. Ther Adv Hematol. 2019 Sep 9;10:2040620719873321
- Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014 Oct 30;124(18):2804-11.
- Dingli D, et al. Clinical Burden of Paroxysmal Nocturnal Hemoglobinuria Among Patients Receiving C5 Inhibitors in the United States. Blood. 2020;136 (S1):2.
- Risitano AM and Peffault de Latour R. How we(‘ll) treat paroxysmal nocturnal haemoglobinuria: diving into the future. Br J Haematol. 2021 Aug 5. Online ahead of print.
- Karadag FK, et al. Is allogeneic stem cell transplantation a good option for paroxysmal nocturnal haemoglobinuria? EMJ Hematol. 2020.
- Garg A, et al. Current status of stem cell transplantation in paroxysmal nocturnal hemoglobinuria. Appl Hematol. 2020;11:161-8.
- PNDS Aplasies médullaires acquises et constitutionnelles. Juillet 2019.
- orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=447*
- Bektas M et al. Paroxysmal nocturnal hemoglobinuria: role of the complement system, pathogenesis, and pathophysiology. J Manag Care Spec Pharm. 2020 Dec;26(12-b Suppl):S3-S8.
Oct., 2023